
By James P Watson with contributions and editorial assistance by Vince Giuliano
This Part 4 blog entry goes even further into the extended NAD story, examining important subtopics:
- The ratio NAD+/NADH in cells and the body, which may be as or more important than absolute levels of NAD+ for driving health.
- The NQ01 gene which drives the NAD+/NADH ratio, and factors related to its activation and expression: BET proteins, the 20S proteasome, BET inhibitors, NQ01 regulation of PGV-1alpha, Nrf2 regulation, etc
- The Warburg effect, changes in cell metabolism characteristic of cancers and aging, its causes, effects and how NAD+ level is only one of several factors affecting it. Why the view that “The Warburg effect is caused by a nuclear state of pseudohypoxia which is caused by insufficient NAD+” is incomplete.
- Reversing Warburg metabolism – known approaches and the possible use of phytosubstances; possible limits of reversal.
- SIRT1 and inflammation, and why control of inflammation has such paramount importance
- In the course of these discussions, a review of possible intervention approaches. ones not commonly discussed in the longevity litterture.
 NAD+ kinase Image source
NAD+ kinase Image source
The Part 1 blog entry in the NAD World series provided an overview treatment of the NAD World and its nuances: to identify the major molecular entities involved, their roles, health and longevity ramifications, the reasons for the current excitement, and to begin to clarify what is actually known and what the remaining uncertainties are.
The Part 2 blog entry in the NAD World series concentrates on the reasons for focusing on NAD+, particularly with respect to interventions that are seriously likely to lead to longer healthier lives. It discusses molecular processes in the NAD salvage cycle that are responsible for the health-inducing and life-extending properties of calorie restriction, and further discuss the key roles of Sirtuins, SIRT1, SIRT6 and SIRT7 in particular.
The Part 3 blog entry in the NAD World series identifies 30 Major Factors that Control SIRT1 Expression, SIRT1 Activity, and SIRT1-mediated Aging.I. The NAD+/NADH ratio and what affects it. The NQ01 gene.
The Part 5 blog entry in the NAD World series is concerned with the conflicting roles of extra-cellular NAMPT, which behaves as both an enzyme and a cytokine, how it is implicated in inducing many disease processes as well as having some positive health impacts.
I. The NAD+/NADH ratio and what affects it. The NQ01 gene
What is of prime importance for health and longevity may not be the actual concentration of NAD+ in cells or cell nucli, but rather the NAD+/NAD ratio which may not be affected by NAD precursor supplements and rather be driven by other matters such as expression of the NQ01 gene,
In the Part 1 blog entry, we characterized the NAD Salvage Cycle by which NAD+ and NADH (the oxidized and reduced forms of NAD) are cycled back into each other and the de-novo pathway via which new NAD is introduced into the body and cells. And. in Parts 1,2 and 3 we discussed how various factors affect the cycling process such as circadian clock gene control. In Part 3 we introduced and initially discussed the NQO1 gene which is a point of departure for the current discussion. “The longevity gene, NQO1, regulates aging by altering the NAD/NADH ratio in cells. NQO1 does this by oxidizing NADH to NAD. Beta-lapachone increases NQO1 enzyme activity and quercetin increases Nrf2-mediated gene expression of NQO1.”
I am now confidence that supplementation with a NAD+ precursor like NMN or NR transiently increased the ratio of NAD+ to NADH (NAD/NADH), but the ratio returns to normal in the course of continued supplementation. This is because there is an enzyme that regulates the NAD/NADH ratio. The ratio is NOT determined by dietary intake or IV intake of any compound. It is enzymatically controlled. That enzyme is called “NAD(P)H dehydrogenase, quinone 1″, or NQO1 for short. NQO1 is an unusual gene in that it requires NADH as a co-factor but does not convert the NADH into nicotinamide, like the Sirtuins or PARPs. Instead, it converts NADH to NAD+. (i.e. it only oxidizes NADH to NAD+).
The oxidation of NADH increases NAD+ levels within the cell and at the same time, decreases NADH levels within the cell. As a result, the NAD+/NADH ratio increases (or the NADH/NAD+ ratio decreases).
Why is NQO1 such an important gene? Well here are the “Top 10 Reasons”.
1, NQO1 regulates the intracellular redox state of the cell and thus, the ratio of NAD/NADH in the cell.
This is the main reason why I now think that NQO1 is so important. Mice that are homozygous negative (both genes knocked out) for NQO1 have an increase in the NADH/NAD ratio (and an increase in the NADPH/NADP ratio). NQO1 knockout mice strangely enough have lower blood glucose levels, less abdominal fat. However, they have higher levels of triglycerides, beta-hydroxybutyrate, pyruvate, and lactate. They also have higher levels of glucagon. More importantly, the NQO1 “knock out” mice have lowered rates of pyridine nucleotide synthesis, reduced glucose metabolism, and reduced fatty acid metabolism. This is not surprising since NQO1 is the controller of the 20S PC mediated degradation of PGC-1alpha.
Unfortunately, cancer cells have also discovered this wonderful property of NQO1. Many cancers unregulate NQO1 either via Nrf2 pathways or by other methods, such as the loss of miR suppression of mRNA for NQO1. A recent study shows that higher levels of the NQO1 protein predict poor prognosis in non-small cell lung cancer. This is a sobering thought – cancer cells up-regulate antioxidant genes! This does not mean that we should avoid phytosubstances that up-regulate NQO1, it just means that “cancer cells are smart”.
Introducing: BET proteins
The opposite problem occurs in aging cells. Aging cells have lower levels of expression of the gene NQO1. This is not just due to a “lack of broccoli” or a “lack of exercise”. Instead, the gene NQO1 is down-regulated by proteins called “epigenetic readers”. The two “epigenetic readers” that suppress the Nrf2-induction of the NQO1 gene are called “Bromodomain and Extraterminal Proteins” (or BET proteins). Specifically, Brd2 and Brd4 proteins “sit on top” of the histone protein acetylated lysines at the promoters of the Nrf2-dependent genes. As a consequence, Nrf2 and the other transcription factors that “turn on” NQO1 gene cannot turn the gene on. (This is why BET inhibitors like JQ1 are so exciting).
Summary: NQO1 regulates the ratio of NAD/NADH and the ratio of NADP/NADPH by oxidizing NADH to NAD+. Warburg-type metabolism ensures that most of the NAD(H) within the cell is in the reduced form (NADH). NQO1 is one of the few genes that oxidizes NADH to NAD+. When both genes for NQO1 are “knocked out”, there is even more NADH in the reduced state. This results in a lower NAD/NADH ratio (or higher NADH/NAD ratio). Thus, NQO1 is the “anti-Warburg gene”. This is why NQO1 is so important.
References:
2015 NQO1 protein expression predicts poor prognosis of non-small cell lung cancers
2013 Bromodomain and extra-terminal (BET) proteins regulate antioxidant gene expression
2. NQO1 regulates the level of the co-activator, PGC-1a,within the cell by controlling degradation rate via the 20S proteasome.
PGC-1a is my “favorite co-activator” because it is so important in mitochondrial biogenesis. I always thought that exercise activated PGC-1a gene via the “exercise kinase” called AMPK. (See he blog enry PGC-1-alpha and exercise). This is why I was shocked to find out that NQO1 actually regulates PGC-1a, not by the increase in expression of the NQO1 gene, but the the rate that PGC-1a is degraded. Expression of NQ01 keeps PGC-1a from being degraded. What I found out is that the level of PGC-1a protein in a cell is primarily determined by its degradation rate, not its synthesis rate.
Like many regulatory factors, PGC-1a has an extremely short half life. All of these extremely short-lived proteins are regulated by degradation rates, not synthesis rates. In the past, it was thought that PGC-1a degradation was only regulated by the ubiquitin-proteasome system (UPS). The UPS method involves a “protein tagger” that goes around putting a ubiquitin “tag” on the protein to be degraded.
Introduce: the 20S proteasome
However, recently a new process of proteasomal degradation has been discovered that does NOT involve any ubiquitination. Specifically, this proteasome does NOT require ubiquitination of the protein and this proteasome system is called the “20S proteasome catalytic particle” (aka 20S PC). Unlike the ubiquitin-dependent, 26S proteasome system (UPS), the 20S proteasome does not require protein unfolding to degrade the protein. (i.e. it can degrade proteins even without unfolding them). Moreover, the 20S proteasome can handle oxidized proteins much better than the UPS 26S proteasome. As a consequence, the 20S proteasome is the “oxidized protein degrader in stressed cells”. For instance, it takes 4 times as much hydrogen peroxide to inhibit the 20S proteasome as it does to inhibit the 26S proteasome of the UPS.
Introducing: intrinsically disordered proteins (IDPs)
Not all proteins are degraded by the 20S proteasome, however. The main type of proteins degraded by the 20S PC system are called “intrinsically disordered proteins” (or IDPs). Interestingly, the 20S proteasome system seems to be regulated by oxidative stress, via the glutathionylation of cysteine residues in the alph-rings of the 20S proteasome.
In conclusion, PGC-1a is a “intrinsically disordered protein” (IDP) that is regulated by its degradation rate. When PGC-1a is damaged by oxidation or when the cell is under oxidative stress (like with aging), the 20S proteasome controls its degradation rate and thus the levels of PGC-1a within the cell. Other IDPs besides PGC-1a include p53, c-fos, C/EBPa, p63, p33, p73a, and ornithine decarboxylase (ODC).
Interestingly, the 20S PC system has a “gate keeper” that inhibits the IDPs from being degraded. Guess who the “gatekeeper” is for 20S PC? Yes, it is NQO1. That is how NQO1 expression keeps PGC-1a around.
Summary:
There is strong evidence now that the levels of PGC-1a in cells is regulated primarily by the degradation rate of PGC-1a, and only secondarily by the gene expression of the PGC-1a gene. There are two degradation pathways for PGC-1a. The two pathways are the Ubiquitin Proteasome system (UPS) and the ubiquitin-independent proteasome system called 20S PC..
Under conditions of no oxidative stress, the UPS system may regulate PGC-1a levels within the cell. However when the cell is under cellular stress and the PGC-1a protein is damaged by ROS-induced oxidation, the 20S proteasome controls the degradation rate of PGC-1a. NQO1 is the “gate-keeper” for this 20S PC system that prevents PGC-1a from being degraded during periods of cellular oxidative stress. Thus with aging, the 20S PC system is more important than the26S proteasome (i.e. the UPS) and thus the 20S proteasome degrades PGC-1a in the cell, unless NQO1 protects it from degradation. Thus it appears that under conditions of oxidative stress, such as with aging, NQO1 may be a major factor that controls the concentration of PGC-1a in the cell.
References:
2013 The Protein Level of PGC-1α, a Key Metabolic Regulator, Is Controlled by NADH-NQO1
2001 Degradation of oxidized proteins by the 20S proteasome
2014 Regulating the 20S Proteasome Ubiquitin-Independent Degradation Pathway
1998 Comparative resistance of the 20S and 26S proteasome to oxidative stress
2013 The Protein Level of PGC-1α, a Key Metabolic Regulator, Is Controlled by NADH-NQO1
2006 20S proteasomes and protein degradation “by default”
1996 Degradation of oxidized proteins in K562 human hematopoietic cells by proteasome
2013 Redox regulation of the proteasome via S-glutathionylation
3. Lower levels of NQO1 leads to increased sensitivity to chemical-induced skin carcinogens.
A very interesting study was done looking at skin-induced cancer from carcinogens. This study looked at NQO1-null mice and found that their levels of NADH was higher (as expected).
Normally, when skin is exposed to chemical carcinogens, p53 is rapidly unregulated. In the NQO1-null mice, exposure to chemical carcinogens did not induce p53 and as a result, the cells did not undergo apoptosis. Instead, they underwent transformation to cancer cells. This is an amazing and very important finding. One feature of p53 induction in the skin is an increase in the appearance of melanin in the skin. This is normally called a “tan” by most people, but on a molecular level, a “tan” is actually p53 induction. This is why people with dark skin have a lower incidence of skin cancer (it is not all to do with the sun block effect of melanin. It is all about p53).
In a study of various mutations in the NQO1 gene in human basal cell carcinomas, 3.1% of 457 cases were found to have “loss-of-function” mutations in the NQO1 gene. Those “NQO1 loss-of-function” individuals were found to have more skin cancers than those with other mutations, but this was not statistically significant. The authors concluded that NQO1 mutations were clearly associated with skin cancer risk, but that these mutations only accounted for a minority of skin cancers.
Summary: NQO1 stabilizes p53 and prevents its degradation. p53 levels in the cell is tightly regulated by two separate degradation pathways – a ubiquitin-dependent pathway that is dependent on the p53 binding partner, Mdm2; or the ubiquitin-independent pathway that is dependent on NQO1. It appears that the NQO1-dependent (ubiquitin-independent) pathway is the most important pathway for regulating p53 levels within the cell.
In the experiment above, NQO1-null mice did not induce p53 in response to carcinogens and the damaged skin cells would not undergo apoptosis, as they should to prevent cancer formation. As a result, the NQO1-null mouse skin cells developed cancer. These effects were thought to be directly due to the lack of binding of NQO1 to p53, which would preven the 20S PC degradation of p53 in the cell. This means that p53 induction and cell apoptosis is dependent on NQO1-mediated stabilization of p53, preventing the 20S PC degradation of p53. This is another reason why HQO1 is so important. However in humans, loss-of-function mutations in the NQO1 gene only account for 3.1% of human skin cancers.
References:
4. NQO1 regulates blood pressureby eNOS, ACE, and an LKB1/AMPK-mediated preservation in GTPCH-1
Another fascinating study showed that activation of NQO1 ameliorates spontaneous hypertension in a rat model. As you may know, spontaneous hypertension does not normally occur in rodents. But in certain strains of inbred rats, bred to develop spontaneous hypertension, high BP does occur and is thought to be mediated by a decline in nitric oxide production by endothelial cells. In this rat model of spontaneous hypertension, activation of NQO1 by beta-lapachone relieved the hypertension in these rats. The positive effects of beta-lapachone were thought to be mediated by NQO1-induction of endothelial nitric oxide (eNOS).
Another study showed that the effect of NQO1 was to regulate the acetylation of eNOS. When an eNOS inhibitor was used, the positive effects of beta-lapachone was completely blocked.
In a separate study, beta-lapachone was used to study the effects of the shedding of the enzyme angiotensin converting enzyme (ACE), which converts Angiotensin I to Angiotensin II in the blood stream. This study showed that beta-lapachone increased NQO1 activity which resulted in reduced cleavage and secretion of ACE into the extracellular space surrounding the cells that synthesized ACE.
In the most recent study, further elucidation of the eNOS mediated mechanism was analyzed and figured out. In this study, they showed that the increase in NAD+ levels in the aortic endothelial cells resulted in an increase in LKBA deacetylation, and AMPK phosphorylation. This was followed by an increase in GTP-cyclohydrolase-1 preservation and tetrahydrobiopterin/dihydrobiopterin ratio. This explained the rest of the story on how beta-lapachone reduced blood pressure.
Both beta-lapachone and the polyphenol, epicatechin, have the effect of reducing blood pressure. Beta-lapachone does this via the direct activation of NQO1, whereas epicatechin does this by activating Nrf2. Nrf2 is the transcription factor that turns on the NQO1 gene.
Summary: NQO1 elevated the ratio of NAD/NADH in the endothelial cells and increases eNOS activity via an AMPK-dependent mechanism. The increase in AMPK phosphorylation resulted in a preservation of the GTP cyclohydrolase-1 (GTPCH-1), which resulted in a lowering of blood pressure. The elevation in the NAD/NADH ratio also results in a reduced cleavage and secretion of ACE into the bloodstream, thereby reducing Angiotensin II formation. As a result of the eNOS-mediated method and the ACE-reduction mediated molecular mechanism, the hypertension in rats resolved. As a result of all this research, NQO1 activation has been recently proposed as a strategy for controlling hypertension (see lst reference below).
References:
2013 NQO1 Activation Regulates Angiotensin–Converting Enzyme Shedding in Spontaneously Hypertensive Rats
2014 Enhanced activation of NAD(P)H: quinone oxidoreductase 1 attenuates spontaneous hypertension by improvement of endothelial nitric oxide synthase coupling via tumor suppressor kinase liver kinase B1/adenosine 5′-monophosphate-activated protein kinase-mediated guanosine 5′-triphosphate cyclohydrolase 1 preservation
2014 NQO1 activation: a novel antihypertensive treatment strategy?
5. “Loss of function” Polymorphisms in the NQO1 gene are associated with carotid artery atherosclerosis/plaques and stroke risk
This was the most amazing study. There is a well-known polymorphism in the NQO1 gene called the “C609T variant.” The C609T polymorphism results in a complete loss of enzymatic activity of NQO1 due to protein instability. The C609T SNP is very common in Asia and has been well-described in Japanese, Korean, and Chinese ethnic groups. Individuals with the “C allele” have a lower risk of carotid plaque disease, whereas individuals with the “T allele” have a higher risk of carotid atherosclerotic disease (OR = 1.65). In Korea, 42% of the population have one “T allele” and 1% of the population have two copies of the “T allele”. 16% of Caucasians have one or two copies of the C609T variant, whereas 49% of Chinese have one or two copies of this SNP.
Another common polymorphism in the NQO1 gene is the C465T mutation. This SNP results in a reduction in enzyme activity, but not a complete loss of function like the C609T SNP. The incidence of this SNP is very low in all populations, varying from 0-5% (see reference below).
References:
2009 The C609T variant of NQO1 is associated with carotid artery plaques in patients with type 2 diabetes
6. Bromodomain and Extraterminal Proteins (BET) supppress Nrf2-mediated gene expression (including NQO1)
Introducing: BET protein inhibitors
We mentioned Bromodomain proteins in Item 1 above. As you may recall, these are “epigenetic readers” that “read” the post-translational modifications of histone proteins. The BET proteins (Brd2, Brd3, Brd4, and BrdT) bind to acetylated lysine residues on histone and non-histone proteins. As a result, they either INCREASE or DECREASE the transcription of the genes associated with these acetylated lysine histones. As it turns out, the Nrf2 genes are regulated by Brd2 and Brd4 proteins. These complex with the acetylated lysine residues on histones located at the promoters of Nrf2-regulated genes. As a result, Nrf2 either cannot increase or decrease the expression of these target genes. When they gave the cells a BET protein inhibitor called JQ1, it increased the expression of Nrf2-genes including HO-1, NQO1, and GCLC, all of which are important in anti-oxidant defense and regulation of intracellular redox state. JQ1 administration resulted in a 10-fold increase in HO-1messenger RNA and a 3-fold increase in HO-1 protein levels. JQ1 administration resulted in a 3-fold increase in NQO1 mRNA expression and a 3-fold change in NQO1 protein expression. JQ1 also increased mRNA expression for GCLC by 2-4 fold and GCLC protein expression by 3-4 fold.
Interestingly, JQ1 also inhibited the expression of the ROS-producing protein, Nox. As a result of this down-regulation of Nox, there was less free radicals in the cell (less H2O2). This resulted in less oxidative stress in the cell due to both a reduced ROS production and increased anti-oxidant enzymes.
This page lists 14 other BET inhibutors, And this 2011 paper discusses the discovery and characterization of small-molecule BET inhibitors
Conclusion: Bromodomain “epigenetic readers” can “shut off” the expression of Nrf2 genes and increase the expression of free-radical producing genes (Nox). Inhibition of BET proteins with JQ1 has the effect of increasing the expression of Nrf2 genes and decreasing the expression of free radical-producing genes (Nox). This may be a key discovery as one of the major causes of oxidative stress-induced aging may be BET proteins.) And may also be why eating Broccoli and exercising have failed to lengthen life span. JQ1, the most well-studied BET inhibitor, suffers from poor pharmacokinetics with a high clearance and low oral biovailabillity in animal studies. This is why a lot of work is going into developing better BET inhibitors.
References:
2013 Bromodomain and extra-terminal (BET) proteins regulate antioxidant gene expression
2014 Bumping into BET inhibitors
2014 New benzazepine BET-inhibitors with improved oral bioavailability
7. The NQO1 gene is regulated by other factors besides Nrf2 – c-Jun, Nrf1, Jun-B, Jun-D, etc.
Quercetin “turns on” the NQO1 gene via Nrf2. Dioxin “turns on” the NQO1 gene via both AhR, Arnt, and Nrf2. Luteolin inhibits expression of NQO1 and drug-metabolizing enzymes via AhR and Nrf2 pathways.
As you may know, the levels of Nrf2 and the location of Nrf2 proteins within the cell is primarily regulated by the binding partner of Nrf2, aka Keap1. There is a new name for Keap1, called INNrf2, but this new name is having trouble getting any attention in the scientific literature This Important transcription factor and its binding protein havr been often discussed in this blog. A comprehensive series of entries on Nrf2 was published in this blog in 2012: Part1, Part2 and Part3.
An older article from 2000 showed that the NQO1 gene is regulated by several factors other than Nrf2 binding to the ARE segment of the NQO1 promoter. They showed the transcription factor, c-Jun, can also bind to the ARE promoter sites on Nrf2-dependent genes. So do the transcription factors Nrf1, Jun-B, and Jun-D. Polyphenols can “turn on” NQO1 via Nrf2. Toxins like dioxin can also ‘turn on” Nrf2, but require the assistance of the aryl hydrocarbon receptor, AhR, and the the aryl hydrocarbon receptor nuclear translocator, Arnt.
References:
2000 Regulation of genes encoding NAD(P)H:quinone oxidoreductases
2001 Induction of human NAD(P)H:quinone oxidoreductase (NQO1) gene expression by the flavonol quercetin
1991 Human NAD(P)H:quinone oxidoreductase (NQO1) gene structure and induction by dioxin
8. The Asian vegetable, “pak choi,” reduces colon inflammation and colon cancer even better than broccoli sulforaphanes
In Asia, there is a very popular type of green, leafy vegetable called “pak choi”. As a child, I ate this frequently as part of our regular diet in Thailand. It is not a particularly “tasty” vegetable and reminds me of a cross between spinach and cabbage, but when boiled or steamed, it is a common vegetable eaten with rice.
A recent study showed that pak choi and brassica vegetables both activated cytoprotective genes, but the sets of genes that were activated were different. Specifically, pak choi, broccoli, brussel sprouts, and other brassica vegetables all activated the typical Nrf2-target genes (NQO1, GSTM1, SRXN1, GPX2), whereas pak choi alone activated the AhR target gene,CYP1A1. The relevance in the current context is that NQO1 belongs to a group of the aryl hydrocarbon receptor (AhR) battery of drug-metabolizing enzymes that are characteristically induced by both AhR agonists and Nrf2 activators(ref). r
More importantly, in the studied mouse models of colitis and colon cancer, the glucosinolate-rich pak choi drastically reduced colitis and colon tumor number, whereas the broccoli-diet did not reduce colitis or colon tumor number in mice.
Conclusion: The presence of glucosinolates (sulphoraphanes, etc.) in food does not necessarily reduce colon inflammation, and colon cancer. It appears that certain foods may have more stable or different glucosinolates that are more effective than others at preventing cancer and inflammation. The Asian vegetable, pak choi, appears to be more effective than broccoli and other brassica vegetables in down-regulating inflammation and preventing colon cancer. (This study was done in Germany, by the way, and was not sponsored by the pak choi industry).
Reference: 2014 Glucosinolates from pak choi and broccoli induce enzymes and inhibit inflammation and colon cancer differently
9. Physical methods and many readily available drugs and phytosubstances increase NQO1 expression or increase NQO1 activity
Including hyperthermia, heat shock, photodynamic herapy, sulindac, dimethylfumarate, taxifolin, sulforaphane, resveratrol, and cisplatin.
A recent discovery that an old, common, generic NSAID that is still available at the drug store also activates the NQO1 gene. Sulindac, a long neglected compound used to treat arthritis, activates the NQO1 gene. Also, previous work has shown that several other compounds up-regulate the NQO1 gene or increase the activity of NQO1. This includes cisplatin, resveratrol, dimethyl fumarate, taxifolin, sulforaphane, and the glucosinolates in pak choi. Since the cancer-killing effects of beta-lapachone are dependent on the levels of NQO1, all of the above compounds work synergistically to kill cancer with beta-lapachone.
Several physical methods have been shown to increase NQO1 gene or protein activity. This includes hyperthermia, heat shock, and photodynamic therapy. This may be how photodynamic therapy works in cancer cells. Interestingly, beta-lapachone works synergistically with these physical methods as well.
References:
2015 NQO1 protein expression predicts poor prognosis of non-small cell lung cancers
2014 The Chemotherapeutic Effects of Lapacho Tree Extract: β-Lapachone
10. Beta-lapachone, a compound found in the bark of the South American Lapacho tree, is a potent activator of the NQO1 protein and produces ROS in cancer cells, but reduces ROS in non-cancer cells. It also inhibits pathological retinal neovascularization, but does not inhibit physiological neovascularization.
The most exciting thing about NQO1 is that there is a natural, cheap, compound found in the tree bark of a South American tree. The compound is called beta-Lapachone and is a NQO1 activator. Specifically, NQO1 is a “two-electron transfer” enzyme that can extinguish free radicals in normal cells, but produces free radicals in cancer cells. It has been shown to be a very effective compound for treating lung cancer. Here is how it works:
- Beta-lapachone undergoes a redox cycle by NQO1, which reduces beta-lapachone to an unstable semiquinone. The semiquinone then rapidly undergoes a two-step oxidation back to the parent stable compound, beta-lapachone. This produces what is called a “perpetuating futile redox cycle”. This results in an unbalance of intracellular reactive oxygen species in cancer cells, resulting in the cell death of the cancer cells. This “perpetual futile redox cycle” is totally dependent on the concentration of NQO1 within cells. Here is a diagram of the reaction:
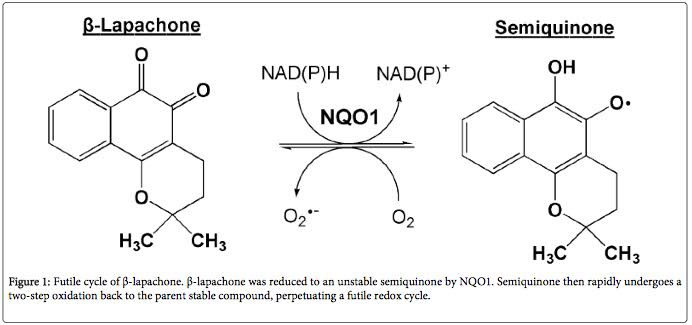
Illustration reference: 2014 The Chemotherapeutic Effects of Lapacho Tree Extract: β-Lapachone
The downstream effects of perpetual futile redox cycling include 4 apoptotic pathways and one necroptotic pathway:
- Mitochondrial-induced apoptosis – The induction of ROS in mitochondria opens the MPTP pores and results in PARP activation and caspace activation. This induces apoptosis.
- ER-induced apoptosis – The induction of ER stress induces sarcoplasmic release of calcium which induces high levels of cytoplasmic Ca++. This also induces apoptosis via the ER.
- DNA-damage mediated apoptosis – beta-lapachone also induces Topoisomerase I and II. The activation of topoisomerases induces DNA breaks, which induces PARPs. This PARP hyper-activation induces apoptosis independently from mitochondrial ROS or ER stress.
- Cell cycle arrest-induced apoptosis – The futile redox cycling of beta-lapachone also induces cycle cycle arrest via the activation of p21, p27, and the phosphorylation of JNK, PI3K, and Akt. This induces cancer cell apoptosis as well.
- Calpain-induced cell necrosis – Unlike the 4 pathways above, futile redox cycling also induces calcium influx into the cells independently of ER stress. This calcium influx into the cell activates Calpain, which induces cell death by the necrosis pathway, not the apoptosis pathway.
Conclusion: beta-lapachone induces cancer cell death by five different pathways, all dependent on perpetual futile redox cycling which is dependent on NQO1 expression. Here is a diagram that illustrates these 5 pathways:
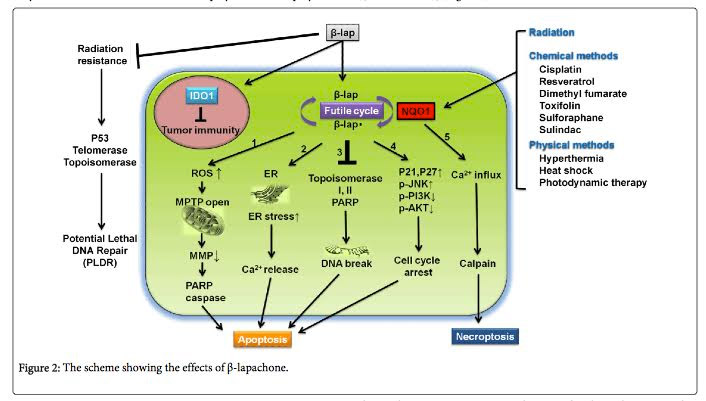
Illustration reference: 2014 The Chemotherapeutic Effects of Lapacho Tree Extract: β-Lapachone
Interestingly, Sulindac, the old generic FDA-approved non-steroidal anti-inflammatory drug, augments the cytotoxicity of beta-lapachone in lung cancer cells. This appears to be due to its effect of up regulating expression of the NQO1 gene. Cancer cells with higher levels of NQO1 actually are more sensitive to beta-lapachone than cancer cells with lower expressions
of NQO1. This may be an “Achilles heel” discovery for cancer cells, allowing us to exploit one of the vulnerable points of cancer cells that have unregulated their anti-oxidant capacity (Many cancers have unregulated Nrf2).
Beta-lapachone shows a lot of promise in treating the most common cause of blindness – neovasculariation of the retina. In the retina, beta-lapachone inhibited pathological retinal neovascularization induced by HIF-1alpha. Unlike VEGF inhibitors that are currently being used for inhibiting retinal neovascularization, beta-Lapachone does not inhibit physiologic angiogenesis…..only pathologic angiogenesis.
References:
2014 The Chemotherapeutic Effects of Lapacho Tree Extract: β-Lapachone
II. The Warburg effects; its causes and consequences and roles in aging in the NAD World

The Warburg effect is an important phenomenon characteristic of aging. It is best known for its existence in cancer cells as well as in aging, and for having several negative biological consequences, though there can be a positive side to it too. The essence of the Warburg effect is a metabolic transformation from energy production from oxidative phosphorylationin the mitochondria to glycolosis. The effect was originally seen in cancer cells. ” Warburg’s hypothesis was postulated by the Nobel laureate Otto Heinrich Warburg in 1924.[3] He hypothesized that cancer, malignant growth, and tumor growth are caused by the fact that tumor cells mainly generate energy (as e.g. adenosine triphosphate / ATP) by non-oxidative breakdown of glucose (a process called glycolysis). This is in contrast to “healthy” cells which mainly generate energy from oxidative breakdown of pyruvate. Pyruvate is an end-product of glycolysis, and is oxidized within the mitochondria. Hence, according to Warburg, the driver of cancer cells should be interpreted as stemming from a lowering of mitochondrial respiration. Warburg reported a fundamental difference between normal and cancerous cells to be the ratio of glycolysis to respiration; this observation is also known as the Warburg effect(ref).” Although much of the research literature related to the Warburg effect is cancer-related, in recent years it is being seen to be very important in aging as well. Specifically, I think there is strong evidence that Warburg metabolism reduces the NAD/NADH ratio in the nucleus, the cytoplasm, and the mitochondria. And, the Warburg effect may be the main reason why that decline is observed in aging.
The Warburg effect and cancers
I don’t think I understood Warburg metabolism until I read the following articles.
2009 Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation
Wikipedia: Warburg hypothesis
2006 Cancer’s Molecular Sweet Tooth and the Warburg Effect
2008 Cellular life span and the Warburg effect
2004 Glycolytic Enzymes Can Modulate Cellular Life Span
2009 Nutrient transporters in cancer: Relevance to Warburg hypothesis and beyond
2006 Cancer’s Molecular Sweet Tooth and the Warburg Effect
2007A High Glycolytic Flux Supports the Proliferative Potential of Murine Embryonic Stem Cells
2009 Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation
2013 Oncogene-induced cellular senescence elicits an anti-Warburg effect
Conclusions:
1. Precancerous cells shift to Warburg type metabolism to achieve immortality. The cost of this shift is a decrease In the NAD+/NADH ratio, which inhibits DNA repair by DNA repair enzymes that are dependent on NAD (PARP, SIRT1, SIRT6) and epigenetic histone modifiers that are dependent on NAD (ARTDs, SIRTs). Also, the previously discussed costs in mitochondrial metabolism which leads to further the Warburg effect.
The poor (slow) DNA repair -allows for DNA mutations to occur at much more rapid rates than what is seen with normal evolution (I.e. Stochastic mutation rates), allowing tumor suppressor genes to be mutated and oncogenes to loose their regulation. The epigenetic dysfunction induced by the change in NAD+/NADH ratios also allow for gene activation or gene silencing to occur by Bromidomain (BET) proteins, histones, DNA methylation, miRNA, and lncRNA over expression. Thus, both DNA mutations and Epigenetic changes occur as a result of the Change in DNA repair that occur with precancerous stages of Cancer.
2. Warburg type metabolism makes precancerous and Cancer cells resistant to oxidative stress because they upregulate gene expression of antioxidant enzymes via Nrf2, Nrf1, c-Jun, FoxO3a, Jun-B, Jun-C, etc. They accomplish this by activating Nrf2 with endogenously produced ROS, which is a potent activator of Nrf2 nuclear localization, as well as Nrf1, FoxO3a, c-Jun, Jun-B, and Jun-C.
3. Warburg type metabolic shifts activate the telomerase gene and cell cycle genes needed for cellular proliferation and survival. This has been well documented in stem cell research. Lowering Oxygen levels in cell culture activates telomerase and cell cycle genes very potently, without the need for TA-65 or any other supplement.
4. An unbiased screen of gene expression in Warburg metabolism cells showed that the “driver” of the Warburg metabolism phenotype is the expression of phosphoglycerate mutase (PGM) and phosphoglycerate isomerase (PGI). SiRNA knockdown of these glycolytic genes induced Cancer cell cycle arrest and cell senescence. Because p53 down-regulates PGM, Cancer cells that survive are the ones that either epigenetically “turn off” p53 or p53 is mutated due to poor DNA repair (see #1 above)
Reference: 2005 Glycolytic Enzymes Can Modulate Cellular Life Span
5. Eating sugar is NOT the cause of Cancer. This is a common fallacy in layperson circles, such as Mercolas Website, etc. Sugar may help spur cancer growth, but the primary contribution of sugar to Cancer cells is that sugar enables mTOR signaling,
6. The last and most important reason that Cancer cells “actively induce the shutting off of mitochondrial function” is that mitochondria are the subcellular organs that induce apoptosis (I.e. Cellular suicide). When mitochondria are “shut off”, you effectively shut off all of the intrinsic suicide pathways in one “fell swoop”! The Caspace and Caspace-independent pathways are all inactivated! Cancer cells call this “A genius maneuver”. We humans call this “Cancer chemotherapy and Radiation resistance” (Cancer cells have a different point of view than we humans do!). Thus the ultimate weapon of the Warburg effect is to inactivate apoptosis!
7. We can possibly counteract the Warburg effect and decline of the NAD/NADH ratio. We can start out to do this by activating NQO1. It will take more than one serving of Broccoli, however. We must address the epigenetic reader problem (Bromidomain proteins 2 and 4 that prevent Nrf2 activation of NQO1 gene expression). See the above discussion in Section I on Brd2 and Brd4. Here are some ideas: In addition to
- upregulating the NQO1 gene by inhibiting Brd2 and Brd4 Bromidomain proteins with BET inhibitors,
- activating perpetual redox cycling with beta-lapachone, and
- inducing Nrf2 with exercise, broccoli, and Pak choi;
there may be some value to trying metabolic inhibitors of Warburg-type metabolism to restore the NAD/NADH ratio to normal. The best established metabolic inhibitors are DCA, 3-BrOP, and 2-DG. These all restore mitochondrial respiration and mitochondrial-mediated apoptosis, thereby stopping cancer cell proliferation and inducing cancer cell apoptosis. There might be equally effective phytosubstance inhibitors. See Section IIIB below, Countering the Warburg Effect with Phytosubstances.
Aging and the Warburg effect
While the Warburg effect has primarily been studied in the context of cancers, it can arise powerfullyin the context of aging. We note first that the effect is not necessarily all-or-none (mitochondrial vs, glycolic metabolism). It can arise gradually over time in the process of aging. Indeed David Sinclair et al’s paper Declining NAD+ Induces a Pseudohypoxic State Disrupting Nuclear-Mitochondrial Communication during Aging identifies a mechanism by which age-related decline of NAD+ leads to mitochondrial dysfunction which leads to the Warburg effect.
In a cell affliced by the Warburg effect, the cell converts most of the NAD+ into NADH with aerobic glycolysis. Two molecules of NAD+ are consumed for every glucose molecule that is oxidized in aerobic glycolysis. Thus you loose two moles of NAD+ for every mole of glucose you consume via the Warburg effect!
The Warburg effect is the use of the glycolytic pathway in the presence of oxygen, whereas anaerobic glycolysis is the glycolytic pathway in the absence of oxygen. Either way, all of the NAD gets converted into NADH with the Warburg effect. Here is a diagram of how/why the Warburg effect lowers NAD+ levels in the cell:
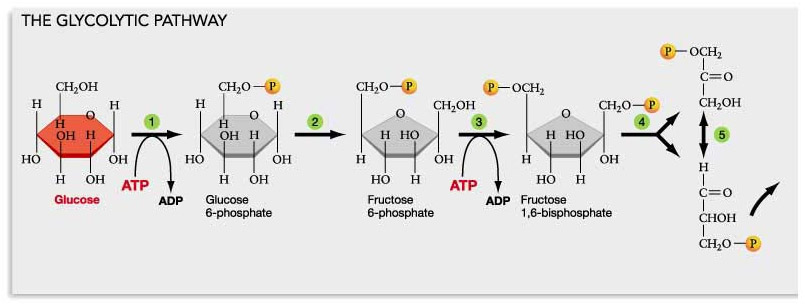
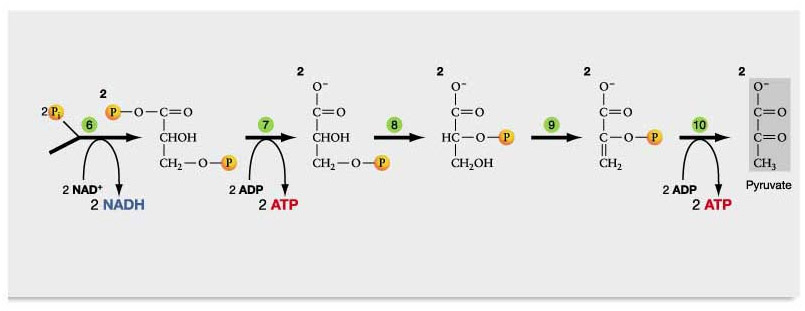
Diagram reference: Glycolysis, Krebs Cycle, and other Energy-Releasing Pathways
Normally, the NADH produced is converted into ATP in the mitochondria. However, when mitochondria do not work properly in aging and with age-related diseases, more and more NADH accumulates within the cell. Some of the NADH is converted to NAD+ when the pyruvate is converted into lactate and then exported to the liver (i.e. the Cori cycle), but the Cori cycle does not keep NAD+ levels high.
Conclusion: Warburg metabolism associated with aging results in low levels of NAD+ within the cell…….this is the culprit! It is also the cause of itself in an unvirtuous cycle
Note that there is a positive feedback loop at work here lowering NAD+: 1. An insufficient level of nulear NAD+ leads to expression of HIF-1alpha. 2. This leads to a pseudohypoxic state that disrupts nuclear-mitochondrial communication leading to deficiency in mitochondrially-encoded proteings resulting into dysfunctional mitochondria. 3. This leads to metabolic reprogramming of the cell to support glycolosis and a Warburg-type metabolism, 4. The Warburg metabolism converts NAD+ into NADH as illustrated in the diagram above. Thus, this cycle of events reinforces itself.
Long non-coding RNAs can induce Warburg-type metabolism
Other actors can promote the Warburg effect. I have now found 3 lncRNAs that induce Warburg-type metabolism as follows:
1. UCA1 (uroepithelial cancer-associated 1) – this is a lncRNA that was discovered in bladder Cancer cells, but appears to be Ubiquitously expressed in other types of cancers and oncancerous cells as well. It upregulates the TOR/STAT3/microRNA-143 pathway, which upregulates the First enzyme in glycolysis, hexokinase 2 (HK2).
Reference: 2014 Long non-coding RNA UCA1 promotes glycolysis by upregulating hexokinase 2 through the mTOR–STAT3/microRNA143 pathway
2. LncRNA p21 – this lncRNA also Induces Warburg-type metabolism by a completely different molecular mechanism. LncRNA p21 interferes with the binding of VHL to HIF-1a. As a result, HIF-1a is not degraded and HIF-1a turns on many glycolytic enzymes, even though there is plenty of oxygen present.
Long noncoding RNA may be the missing “dark matter” that explains the unaccounted for risk of developing cancer, disease, and even aging itself. There appears to be almost 60,000 of these “genes” hidden in the noncoding portion of the human genome, which accounts for 98% of our DNA. LncRNA-p21 is one of the few that has been characterized and may be the “unexplained dark matter” of the Warburg effect in cancer and in aging.
Until recently, long noncoding RNA were not recognized as being major regulators of gene expression. With the recent advent of next generation RNA sequencing (RNA-seq), almost 60,000 long noncoding “genes” have been found in the human genome. As of yet, only a handful of these have been characterized for their functional significance, such as HOTAIR, MALAT1, H19, KCNQOT1, ANRIL, etc.
One of these is “Long noncoding RNA at the p21 locus”, or lncRNA-p21, which is an independent gene downstream from the protein-coding gene, CDKN1A, or p21. When the CDKN1A gene is transcribed, the lncRNA-p21 is also transcribed. Thus although this gene is a separate gene, it is co-expressed with its protein-coding gene.
The lncRNA-p21 does not appear to regulate the CDKN1a gene, however. Instead, it appears to be a lncRNA with wide-ranging effects, regulating distant genes via “trans-regulatory” mechanisms. LncRNA-p21 is a hypoxia-inducible gene. More-over, HIF-1a is responsible for the hypoxia-inducible expression of lncRNA-p21.
In addition, lncRNA-p21 then stabilizes HIF-1a by disrupting the binding of HIF-1a to VHL. The net effect is a “positive feedback cycle“, where HIF-1a induces lncRNA-p21, which then prevents the degradation of HIF-1a by VHL. This “positive feedback cycle” may be the greatest factor that determines cancer formation.
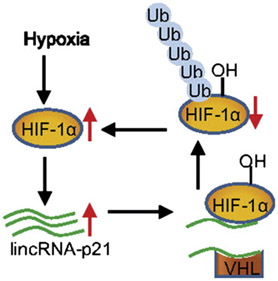
References:
2014 Reciprocal Regulation of HIF-1α and LincRNA-p21 Modulates the Warburg Effect (agstract)
3. CRNDE – this lncRNA name is an Acronym for “Colorectal neoplasia differentially expressed” lncRNA. As expected, it was discovered in colon cancer cells, but like UCA1 and lncRNA p21, appears to be universally expressed in many cancers and noncancerous tissues. There are many splice variants of the CRNDE lncRNA, but the key splice variants include a specific sequence in intron 4 called the “gVC-ln4” sequence. gVC-ln4 copies of CRNDE induce Warburg type metabolism. Interestingly, CRNDE is regulated by insulin signaling. Thus, CRNDE may be the molecular mediator of why the Insulin/IGF-1 pathway causes aging (as opposed to sugar being the cause of aging).
Reference: 2014 CRNDE, a long non-coding RNA responsive to insulin/IGF signaling, regulates genes involved in central metabolism
Conclusions: At least 3 lncRNAs have now been discovered that explain why both aging and Cancer exhibit Warburg-type metabolism. There may well be more such, given that the “Wild West” of long non-coding RNAs is only now being explored. The evolutionary selection pressure for the development of UCA1, lncRNA p21, and CRNDE all are probably for cellular survival or organismal survival. These lncRNAs are one of the farthest “upstream “causes” of aging and cancer that I have seen.
About HOTAIR
Can we identify interventions which can affect the expression of such IncRNAs? I think the answer is Yes. At least this is the case for one non-coding RNA for which expression can be altered by curcumin. This lncRNA is called “HOTAIR“ and is a critical regulator of cancer metastasis and cancer survival. HOTAIR is over-expressed by 100-fold in breast cancer cells and the level of its over-expression correlates with both metastasis and survival in breast cancer. Although work in this area has just begun, one report already shows that the metastasis-preventing effects of curcumin in renal cell carcinoma (RCC) may be mediated via HOTAIR. No other lncRNA has been linked to polyphenol mechanism of action to date, however. Because knowledge of what IncRNAs do is unfolding so rapidly now, we will not be surprised to hear soon of other examples.
Reference: Influence of Curcumin on HOTAIR-Mediated Migration of Human Renal Cell Carcinoma Cells
III Metabolic reprogramming from the Warburg Effect
A. Cells can be weaned off from Warburg metabolism back to mitochondrial respiration via DCA and TMZ. (and off Glutaminolysis with Arsenic Trioxide)
I am fascinated with the idea of using safe, nontoxic compounds for reprogramming our cells to be “weaned off” of aerobic glycolysis and to switch them back to glucose oxidation. I am now convinced this can be done with plant polyphenols, assuming they are effectively delivered tp cells. First, I focus focus on two drugs that are well known to do this, DCA and TMZ. Then I discuss the potential use of plant polyphenols in Subsection B to follow.
DCA and TMZ have been shown to have amazing effects on the heart, especially for heart failure with preserved ejection fraction (HFPEF). However, in addition to treating HFPEF, these compounds appear to have great potential for treating or preventing cancer. Here is what I have learned so far about them.
1. Systemic Factors that “drive” the development of the heart’s dependence on free fatty acids (paradoxic effect….opposite of a ketogenic diet)
Cardiac-intrinsic causes of HFPEF
With diabetes, there is a 2-fold higher risk of heart failure in men and a 5 fold higher risk of heart failure in women. This cannot be explained by advanced glycation end products, coronary artery disease (and MI), and hypertension alone. There are also “cardiac intrinsic mechanisms” that are at work which are responsible for this dramatic increase in HF in diabetics. In addition, non-diabetics develop a type of heart failure where there is no pre-existing heart attack and even in some cases, no hypertension. This is called “normal ejection fraction heart failure”, or HFPEF.
An article on this is: 2003 Heart Failure – The frequent, forgotten, and often fatal complication of diabetes
Metabolic Reprogrammiong in the Heart – “mhs isoform switching”
With HFPEF in diabetes and with aging, there appears to be a metabolic energy substrate switching in the heart from the use of glucose as fuel to the use of free fatty acids as fuel. This gene expression change is referred to as “mhc isoform switching”. Here there is an increase in myosin V3 gene expression and the sarcoplasmic reticulum Ca++ pump ATPase genes. This results in cardiac hypertrophy and metabolic reprogramming. In addition the decreased glucose availability in the heart that occurs with insulin resistance and a high fat diet in diabetes drives this process. As a result, gfat2 is expressed instead of gfat1. Another systemic factor besides glucose and high free fatty acids is the stress hormones produced from the HPA axis and the sympathetic nervous system (SNS). Specifically, the epinephrine (from the adrenal gland) and norepinephrine (from the sympathetic nervous system) that is produced when we are psychologically stressed, and with normal everyday waking hours (i.e. HPA axis hormones and SNS stimulation). In addition to triggering genes that switch fuel use (metabolic reprogramming), the SNS and HPA axis triggers gene expression of “fetal gene programs”. Interestingly, dietary medium chain triglycerides (MCT oil) can prevent this occurrence.
Here are some articles on this:
2007 Proposed Regulation of Gene Expression by Glucose in Rodent Heart
1982 Cardiac alpha- and beta-adrenergic receptor alterations in diabetic cardiomyopathy
2. Systemic Factors that “drive” cardiac fibrosis, cardiac remodeling, and age-related heart failure with preserved ejection fraction (HFPEF)
With diabetes and with aging, there is a second factor that is responsible for the cardiac remodeling that occurs with HFPEF. This is the renin-angiotensin system (RAS), which triggers the expression of extracellular matrix genes (collagen genes and MMP genes), which cause the stiffness of the heart with this disease. Fortunately, there is evidence that this cardiac remodeling is reversible. Ironically, the extract from broccoli, brussel sprouts, cabbage, and cauliflower, Indole-3-carbinol, can trigger this cardiac remodeling! In the study below, withdrawal of I3C reversed the effects of the cardiac remodeling caused by the RAS system. This is very puzzling.
Here are some articles on this:
htt2013 Cardiac remodeling during and after renin–angiotensin system stimulation in Cyp1a1-Ren2 transgenic rats
2007 The Renin-Angiotensin Aldosterone System: Pathophysiological Role and Pharmacologic Inhibition2012 Reversible cardiac remodeling after renin-angiotensin system stimulation in CYP1A1-Ren2 transgenic rats
3. Once cardiac remodeling induces heart failure, ACE inhibitors, beta-blockers, and Angiotensin II receptor blockers do not reverse the heart failure.
Here are some articles on this:
2008 Advances in the treatment of heart failure with a preserved ejection fraction
4. Even after heart failure has occurred with HFPEF, Cardiac Metabolic Reprogramming with DCA or TMZ can dramatically improve cardiac index and improve mechanical efficiency of the heart
Dichloroactetic acid, a simple, cheap compound that is well absorbed orally, induces a “metabolic switching” of cardiomyocytes from the utilization of fatty acids to the utilization of glucose. This glucose utilization is NOT aerobic glycolysis (i.e. the Warburg effect), but instead is true “glucose oxidation” in the mitochondria. This increases the amount of ATP that can be generated and reduces the workload of the heart to generate ATP. The dose is relatively high (50 mg/kg body weight in humans). This means for an average person, they would need to take 3-4 grams of DCA.
Here are some articles on sodium dichloroacetate. References:
2010 Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC
There is a second compound that is a drug widely available in Europe (and Canada) but not in the US. In fact, it is widely available in over 80 countries. This drug is called “trimetazidine”, or TMZ, an is another “metabolic reprogrammer” for the heart. It also makes the heart switch from utilizing free fatty acids to utilizing glucose. DCA and TMZ do not carry out “metabolic reprograming” the same way, however. TMZ does this by inhibiting long chain 3-ketoacyl coenzyme A thiolase, whereas DCA targets Pyruvate dehydrogenase kinase (PDK) by acting as a “pyruvate mimetic”. PDK is an inhibitor of Pyruvate dehydrogenase (PDH). By inhibiting PDK, PDH is maintained in its active catalytic form and mitochondrial pyruvate consumption is increased. Whereas DCA has to be taken in large doses, TMZ is only a 20 mg three times per day dosage and has been used in Europe for over 40 years. It does have one significant rare side effect of causing drug-induced Parkinsonism.
Reference: 2006 Clinical Trial of Trimetazidine , a Partial Free Fatty Acid Oxidation Inhibitor, in Patients With Heart Failure
5. Metabolic reprogramming can also have wonderful anticancer effects by inhibitingWarburg metabolism, inducing differential apoptosis in cancer cells vs normal cells (preserved)
Warburg metabolism is seen in about 90% of cancers. The remaining ones either utilize the glutaminlytic pathway to generate ATP, or still use free fatty acids or glucose oxidation. However, it is thought that more than 99% of cancer cells either use Warburg type metabolism or Glutaminolytic metabolism. There is strong evidence that DCA alone or DCA in combination with genotoxic drugs can make a huge difference in cancer cells that are dependent on aerobic glycolysis. Because cancer cells have been “hardwired” to undergo aerobic glycolysis (Warburg metabolism), it is very difficult to get them to change their metabolism. There are several reasons for this metabolic “hardwiring” in cancer cells. The obvious one is that cancer cells have lots of mitochondrial mutations so they cannot generate the components of electron transport/oxidative phosphorylation that are expressed only in mitochondrial encoded genes (mtDNA). A second reason is that cancer cells have unregulated or stabilized HIF-1a, which encodes for all of the glycolytic enymes. This up regulation of HIF-1a is due to the Insulin/IGF/PI3K/Akt/mTOR pathway being unregulated, the C-myc pathway being unregulated, and the ROS oncogenic pathway being unregulated. The third reason is the loss of p53 gene expression, either due to mutations or due to epigenetic silencing in the p53 gene promoter by CpG methylation and histone-based silencing.
In summary, there are so many overlapping reasons why cancer cells display “Warburg Metabolism” that this form of metabolism appears to be “hardwired” and not changeable by dietary factors, oxygen, fasting, drugs, exercise, or stress reduction. In other words, when it comes to cancer cell metabolism, if they have Warburg-like metabolism, they are stuck with it. And the good news is that this is not necessarily so for normal cells.
DCA has been shown to act as a “pyruvate mimicker” and targets pyruvate dehydrogenase kinase (PDK). PDK normally inhibits PDH. When DCA inhibits PDK, PDH continues to consume mitochondrial pyruvate, which then metabolically reprograms” the mitochondria to utilize glucose for generating ATP, rather than utilizing free fatty acids for generating ATP (i.e. beta fatty acid oxidation). This results in a reduction in serum lactate. This is why DCA is used to treat congenital lactic acidosis. Interestingly, DCA induces apoptosis in cancer cells but does not induce apoptosis in normal cells.
There are now numerous reports from cancer laboratories that show amazing benefits of DCA in experimental models. There is also anecdotal evidence from patients who have self-medicated themselves with DCA and had long term survival associated with with daily use of DCA. The main side effect of DCA is peripheral neuropathy. This side effect can be prevented by keeping the dose below 10 mg/kg/day and by taking oral Thiamine, NAC, and CLA.
References:
2008 Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer
Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC
DCA Site: Cancer papers by Archer and Michelakis
2008 Pyruvate kinase M2 is a phosphotyrosinebinding protein
2008 The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth
2011 Targeting metabolism with arsenic trioxide and dichloroacetate in breast cancer cells
6. Glutaminolysis – the “black sheep” metabolic pathway of cancer cells that is often forgotten.
Anther key metabolic dysfunction in cancer cells is the utilization of glutamate as an alternative energy source. Although this does not occur in all cancer cells, it occurs in chemotherapy-resistant tumors. For instance, promyeloid leukemia and certain subsets of pancreatic cancer utilize the glutaminolytic pathway to generate ATP. There is a very old “metabolic inhibitor” of the cancer cell’s glutaminolytic pathway of generating ATP – Arsenic Trioxide (ATO). ATO is very cheap but hard to get. Alone, it has amazing but toxic effects on certain cancers. When used in combination with DCA, it has a synergistic effect, inhibiting cancer even more than DCA or ATO can do alone.
References:
2011 Targeting metabolism with arsenic trioxide and dichloroacetate in breast cancer cells
2002 Molecular targets of arsenic trioxide in malignant cells
2002 Mechanisms of action of arsenic trioxide
2010 Genistein synergizes with arsenic trioxide to suppress human hepatocellular carcinoma
2009 A novel combination therapy with arsenic trioxide and parthenolide against pancreatic cancer cell
Conclusions: Forced metabolic reprogramming may be a novel strategy for cancer cell apoptosis and for age-related heart failure with preserved ejection fraction. Although little has been done in this arena, I believe that this would be a huge opportunity to explore new ways of attacking aging phenotypes in many organs – heart, skeletal muscle, liver, etc. Unfortunately, little has been done in this area in the anti-aging research field.
B Countering the Warburg Effect with Phytosubstances
Here are some natural compounds I have found that may inhibit aerobic glycolysis:
- Green tea (EGCG) inhibits glycolysis – Inhibits the 6th step of glycolysis at the enzyme Glyceraldehyde-3-phosphate dehydrogenase (GAPDH). EGCG from green tea undergoes “auto-oxidation”, forming “electrophilic quinones” inside cells. This EGCG-quinone fits into the active enzyme pocket of the 6th enzyme in glycolysis, called Glyceraldehyde-3-phosphate dehydrogenase (GAPDH). In the enzyme pocket of GAPDH, the EGCG-quinone then forms a covalent bond with a cysteine thiol amino acid on GAPDH. Mutatgenesis studies that substituted this cysteine with another amino acid eliminated the inhibitory effect of EGCG on GAPDH enzyme activity.
The inhibition of GAPDH by the EGCG-quinone is irreversible, which means that the cell has to synthesize more GAPDH to continue with aerobic glycolysis activity (i.e. Warburg-type metabolism).
Reference: 2008 Covalent modification of proteins by green tea polyphenol (-)-epigallocatechin-3-gallate through autoxidation
Conclusion: Oxidized EGCG, referrred to as “EGCG-quinone”, forms a covalent bond with a cysteine amino acid side chain in the active enzyme pocket of GAPDH, an important enzyme in the glycolytic pathway. Because this covalent bond formation results in irreversible inhibition of the GAPDH enzyme, this is one molecular mechanism by which green tea can reduce or reverse Warburg-type metabolsim.
- Green tea (EGCG) inhibits the conversion of pyruvate to lactate – the conversion of pyruvate to lactate by LDH and the export of lactate to the plasma for hepatic conversion to glucose are key metabolic alterations that allows cells to survive Warburg-type metabolism. EGCG from green tea undergoes “auto-oxidation” and then inhibits another key enzyme in Aerobic glycolysis – LDHA. Lactate dehydrogenase (LDH) has two isoforms, LDHA and LDHB. LDHA is not under the transcriptional control of HIF-1a. LDHA can be inhibited by oxamate, a specific inhibitor of this isoenzyme. As a result, less lactate is produced and the Warburg-type metabolism is disrupted in cancer cells. As a consequence, pyruvate accumulates in the cancer cells and can induce cancer cell death or inhibit cancer cell growth. In the study quoted below, EGCG was found to have the same inhibitory effect on LDHA that oxamate produces.
Reference: 2014 Metabolic consequences of LDHA inhibition by epigallocatechin gallate and oxamate in MIA PaCa-2 pancreatic cancer cells
Conclusion: The inhibition of LDHA is a second molecular mechanism by which oxidized EGCG can inhibit Warburg-type metabolism.
- Green tea (EGCG) inhibits HIF-1a, which would decrease Warburg-type metabolism– Although there are a few studies (two of them) which suggest that catechins from green tea increase HIF-1a stability, most studies have come to the opposite conclusions. Below are some of them:
- EGCG has been shown to inhibit hepatocellular carcinoma by inhibiting VEGF/VEGFR axis (see below).
- EGCG has been shown to inhibit colorectal cancer by inhibiting the VEGF/VEGFR axis (see below)
- ECGC has been shown to inhibit HIF-1a and VEGF in human cervical cancer cells and in human hepatoma cells.
- EGCG from green tea was studied to see what effects the EGCG polyphenol would have on HPV-infected cells. Cells infected with HPV produce oncoproteins E6 and E7, which induce Warburg-type metabolism and cancer formation. This is one of the molecular mechanisms by which HPV infection can cause cancer – by activating Warburg-type metabolism and angiogenesis.
- They found that EGCG inhibited angiogenesis in these HPV cells, both in vitro and in vivo, and also inhibited HIF-1a protein expression in HPV cells.
- EGCG also reduced the secretion of VEGF and IL-8 from these HPV-infected cells. In this same study, however, EGCG had no effect on inhibiting HIF-1a expression.
Several similar studies are listed below. References:
Conclusions: EGCG inhibits HIF-1a, VEGF, and IL-8 production. And oh yes, in addition to all of the above, lets not lose sight of the fact that EGCC promotes the expression of Nrf2 which is going to activate the NQ01 gene which will tend to normalize the NAD+/NADH ratio which, as pointed out earlier, will work against the Warburg effect. .
- Resveratrol inhibits HIF-1a and VEGF expression-here is an example of another natural compound that inhibits Warburg-type metabolism via HIF-1a. Resveratrol is a stilbene found in red wine, red grape skins, Itadori tea, and many other natural compounds. Resveratrol was shown in this study to have major effects on both baseline HIF-1a protein accumulation as well as hypoxia-inducible HIF-1a protein expression. Unlike some of the other compounds listed here, the molecular mechanism for resveratrol-induced decrease in HIF-1a protein appears to be due to an increase in the degradation rate of HIF-1a by the 26S proteasomal unit, not a decrease in the gene expression of HIF-1a. Thus, there should be a synergistic effect of combining a compound like resveratrol with some of the other compounds that inhibit gene expression of HIF-1a
References:
Conclusion: Resveratrol reduces HIF-1a by increasing its degradation rate of HIF-1a by the ubiquitin proteasomal system (UPS), not by down regulating its gene expression.
- Curcumin directly inhibits HIF-1a and VEGF by down regulating their gene expression
There are clearly multiple mechanisms of action for natural products. Curcumin, the active ingredient in turmeric, derived from the roots of the turmeric plant is the main active ingredient in curry spice. Here is the first of two mechanisms that may have anti-Warburg metabolism effects in human cells:
Curcumin has been shown to have major effects on tumor suppression, but all of the mechanisms have not been fully elucidated. In the study below, curcumin was shown to have a direct effect on down-regulating gene expression for HIF-1a. Curcumin also down-regulated gene expression for Vascular endothelial growth factor, VEGF, the downstream target gene of HIF-1a. This effect occurred both under hypoxic conditions (normal anaerobic glycolysis) and under normoxic conditions (Warburg metabolism).
Reference: 2oo6 Curcumin inhibits hypoxia-induced angiogenesis via down-regulation of HIF-1
Conclusions: Curcumin shouild have major direct effects in inhibiting or reversing Warburg-type metabolism, both under hypoxic and normoxic conditions
- Curcumin indirectly inhibits HIF-1a by inhibiting STAT3– Although curcumin has major anti-inflammatory effects due to its ability to inhibit NF-kB signaling, not as much is known about its ability to inhibit Warburg-type metabolism. However, it may do this via STAT3. Curcumin is an inhibitor of the pro-inflammatory transcription factor,Signal transducer and activator of transcription 3, aka STAT3/ STAT3 is a central regulatory of tumor metastasis, but plays an integral role in inducing Warburg metabolism due to its effects on activating HIF-1a. Curcumin inhibits STAT3, which then inhibits HIF-1a, which is a down stream target gene of STAT3. Thus, curcumin indirectly affects aerobic glycolysis or Warburg-type metabolism via STAT3 inhibition.
Reference: 2009 STAT3 as a Central Regulator of Tumor Metastases
Conclusions: Curcumin should have major indirect effects on Warburg-type metabolism via STAT3 inhibition.
- Vitexin directly inhibits HIF-1a protein and also down regulates HIF-1a and VEGF gene expression– this interesting natural flavanoid has a dual mechanism of action on reducing HIF-1a activity. Vitexin is a flavanoid found in many different plants, including Anthurium versicolor(Aquino et al., 2001), Ficaria verna Huds. (Ranuncu- laceae) (Tomczyk et al., 2002), and Cucumis sativus L. (Cu- curbitaceae) (McNally et al., 2003). It has also been shown to be present in Kombucha tea. It was shown to be a potent inhibitor of HIF-1a protein activity in phenochromcytoma cells. Others showed it to be a potent inhibitor of hepatocellular cancer cells via HIF-1a.
References:
2006 Vitexin, an HIF-1α Inhibitor, Has Anti-metastatic Potential in PC12 Cells (abstract)
2006 Vitexin, an HIF-1α Inhibitor, Has Anti-metastatic Potential in PC12 Cells (PDF full text)
2007 Hypoxia and hepatocellular carcinoma: The therapeutic target for hepatocellular carcinoma
IV. SIRT1 and inflammation
NAD+ and SIRT1 availaility play key roles in both acute and chronic inflammation. The 2013 publication Deacetylation by SIRT1 Reprograms Inflammation and Cancer summarizes key issues in the SIRT1-inflammation story. The abstract reports: “NAD+-dependent deacetylase SIRT1 is a master regulator of nucleosome positioning and chromatin structure, thereby reprogramming gene expression. In acute inflammation, chromatin departs from, and returns to, homeostasis in an orderly sequence. This sequence depends on shifts in NAD+ availability for SIRT1 activation and deacetylation of signaling proteins, which support orderly gene reprogramming during acute inflammation by switching between euchromatin and heterochromatin. In contrast, in chronic inflammation and cancer, limited availability of NAD+ and reduced expression of SIRT1 may sustain aberrant chromatin structure and functions. SIRT1 also influences inflammation and cancer by directly deacetylating targets like NFκB p65 and p53. Here, we review SIRT1 in the context of inflammation and cancer.” Five diagrams in that publication lay out the story.
- There are Two Types of Inflammation: Acute and Chronic
According to the article, chronic infections contribute up to 20% to cancer, inflammatory diseases contribute up to 20% of cancer causation, and obesity contributes up to 20% of cancer causation.
- Chronic infection, inflammatory disease, and obesity all produce extracellular signaling compounds that are Toll-like receptor activators, or cytokines, or ROS, or RNS.
- Whereas infections and obesity have other signaling mechanisms, TLR and cytokine signaling are the principle mediators of inflammation and trigger inflammation via three pathways:
- STAT1– This pathway is activated by the IFN family of cytokines. IFN induces STAT1phosphorylation, which results in cytoplasmic-to-nuclear translocation of STAT1, where STAT1 binds to and turns on inflammatory genes. Unlike acetylation of NF-kB, the acetylation of STAT1 inactivates the transcription factor, whereas HDAC3s (not SIRT1) deacetylate STAT1 and thereby allows for phosphorylation and inflammatory gene activation. This process has nothing to do with NAD or SIRT1
Reference: A phosphorylation-acetylation switch regulates STAT1 signaling
How to “fix” this problem: take a polyphenol that is an HDAC inhibitor (Ex: EGCG), also take a STAT1 inhibitor, and reduce IFN signaling
- STAT3– This pathway is activated by the IL-6 family of cytokines (IL-6, IL-10, etc.) IL-6 induces the Janus kinase, called JAK in the cytoplasm, which then phosphorylates STAT3.(This is why it is called the “JAK/STAT3 pathway”). Phosphorylation of STAT3 by JAK results in cytoplasmic-to-nuclear translocation of STAT3, where STAT3 can bind to gene promoters to produce a transient activation of inflammatory genes. IL-10 also activates STAT3, but paradoxically seems to have opposite effects. IL-10 activation of STAT3 produces a sustained expression of genes that are mostly anti-inflammatory. Thus STAT3 signaling can be pro or anti-inflammatory.
HDACs deacetylate STAT3 and disrupt the JAK/STAT3 signaling pathway. These are not NAD-dependent deacetylases, however.
References:
STAT3 activation in response to IL-6 is prolonged by the binding of IL-6 receptor to EGF receptor
Cytokine response is determined by duration of receptor and STAT3 activation
How to “fix” this problem: take a polyphenol that is an HDAC inhibitor (Ex: EGCG), also take a STAT3 inhibitor, and reduce IL-6 signaling
- NF-kB– This pathway is activated by TLR signaling (double stranded RNA, single stranded RNA, bacterial flagellin, bacterial and viral CpG motifs, malarial pigment hemozoin, lipopeptides, lipoproteins, hyaluronan breakdown products such as short hyaluronic acid chains, endotoxin (LPS), amyloid-beta 42, viral proteins, and many other extracellular proteins). TLR signaling phosphorylates the binding partner of NF-kB, called IKB-alpha.
The phosphorylation of IKBa results in the degradation of IKBa, which leaves NF-kB free to translocate into the cell nucleus. NF-kb then binds to gene promoters of the inflammatory gene network. Part of this includes the activation of the NAMPT gene, which increases NAD levels in the acute phase of inflammation. The NAD stabilizes SIRT1a nd activates SIRT1. Thus there is an increase in SIRT1 activity with acute inflammation.
Reference: 2010 TLR-signaling Networks: An Integration of Adaptor Molecules, Kinases, and Cross-talk
SIRT1 activity increases as acute inflammation evolves. The following diagram illustrates the cascade of events that occurs with TLR signaling:
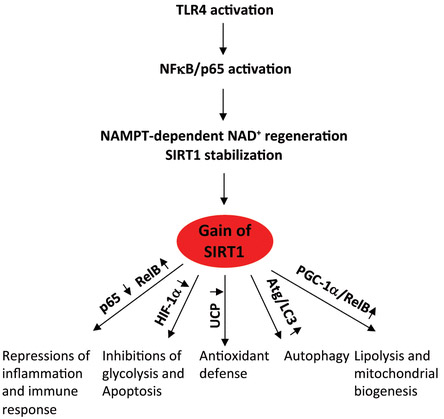
Reference for diagram: Deacetylation by SIRT1 Reprograms Inflammation and Cancer
“Gain of SIRT1 functions during acute inflammation. TLR responses increase NAMPT-dependent NAD+ regeneration and activate SIRT1, which represses inflammation, glycolysis, and apoptosis and increases lipolysis, mitochondrial biogenesis, autophagy, and antioxidants. This sequential process restores homeostasis.”
Whereas bacterial infections, viral infections, surgery, accidents, and other triggers induce acute inflammation, chronic inflammation is typically triggered by disease and aging. For instance, obesity induces chronic inflammation which induces a number of inflammatory diseases, including HTN, type II diabetes, cancer, and atherosclerosis.
Chronic Inflammation produces a different form of gene expression than acute inflammation, however. The differences in acute vs chronic inflammation includes different forms of chromatin (euchromatin vs heterochromatin). The different “chromatin signatures” for each type of inflammation are shown below:
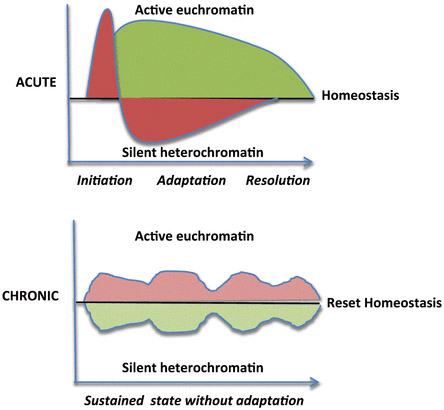
Illustration Reference: Figure 2. Deacetylation by SIRT1 Reprograms Inflammation and Cancer
“Inflammation phenotypes. Acute inflammation modifies the chromatin structure to switch from initiation to adaptation and resolution. Chronic inflammation sustains proinflammatory chromatin.”
With chronic inflammation, several things occur connected with SIRT1. First of all, a high fat diet cleaves SIRT1 and low NAD levels reduce SIRT1 synthesis (see diagram below and article reference for diagram for more info on this).The net result of a high fat diet and lower NAD levels is a loss of SIRT1 function. This results in hyperacetylation of the p65 subunit of NF-kB, which produces pro-inflammatory products. These proinflammatory mediators have a “positive feedback” on the formation of obesity, diabetes, and aging.
The direct downstream effects of reduced SIRT1 include an increase in adiponectin, a decrease in PPARalpha, an increase in insulin and a lowering of UCP2, as well as an increase in telomerase expression, a decrease in UCP2 expression, and a decrease in FoxO1 and FoxO3 mediated pro-survival gene expression.
Reference for diagram below: Deacetylation by SIRT1 Reprograms Inflammation and Cancer
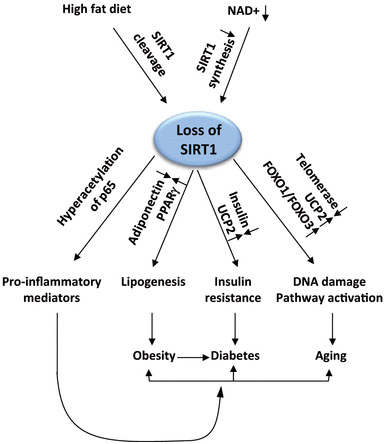
Figure 4.
“Loss of SIRT1 functions during chronic inflammation. High fat diet reduces NAD+ availability and deactivates SIRT1, which promotes inflammation, lipogenesis, insulin resistance, and DNA damage. This unadaptive process prevents a return to homeostasis.”
When it comes to Cancer, SIRT1 has been a major puzzle. Well the puzzle has now been solved – SIRT1 is both a tumor suppressor and a tumor activator. Here is a diagram that illustrates that:
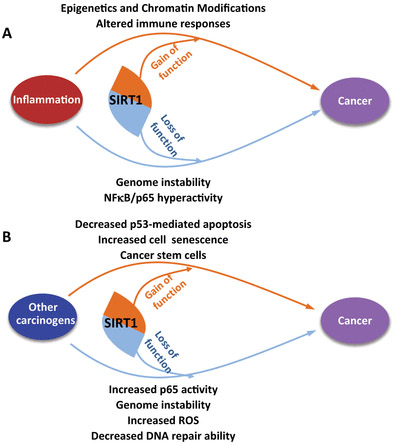
Diagram reference: Figure 5. Deacetylation by SIRT1 Reprograms Inflammation and Cancer “Dual effects of SIRT1 on cancer: (A) inflammation and (B) modifying specific proteins.”
There is a diagram (below) that summarizes the factors that affect SIRT1 gene expression.
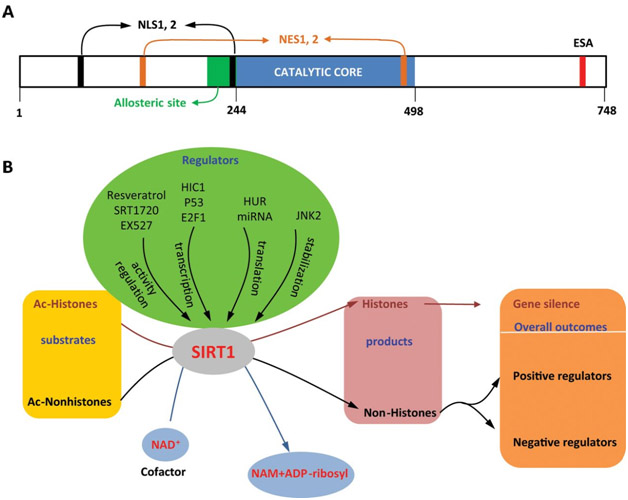
Diagram reference: Deacetylation by SIRT1 Reprograms Inflammation and Cancer Figure 1.
“SIRT1: (A) structure and (B) functions and regulation.”

What is the main supplement for Beta-lapachone? When I search for supplements containing south American tree bark for lapacho tree the only related supplement/cource I can buy is Pau d’Arco. Is this essentially the same thing? Can you comment on this and what daily dosages you have seen that seem reasonable or that you take yourself?
Thank you, sincerely, (as always for an amazing blog)- cam
Hi cam
I do not know the answers to your questions but I think they are good ones. Perhaps we can get Jim to weigh in on this. I too am interested in the personal possibility of supplementing with Beta-lapachone and probably will get around soon to looking into your questions. Vince
Great read over and over!
Thank you!
Thank you for sharing your work and ideas with us. There were indications that some were supplementing with NAD. If I understand the implications of the following from above correctly, are the personal opinions currently that continued NAD supplementation is likely not helpful… or that NAD should possibly be “cycled” [if so, any ideas for cycling?]? I am among those who have been giving it a try [via NR] and certainly haven’t noticed any negative effects.
From above: “I am now confident that supplementation with a NAD+ precursor like NMN or NR transiently increased the ratio of NAD+ to NADH (NAD/NADH), but the ratio returns to normal in the course of continued supplementation”
Best regards,
Jon
Jon
Thank you for this intelligent comment. Speaking directly of personal experience:
1. Narrowly speaking, consuming an aqueous solution of NAD seem to make a significant difference for me for about two weeks and then the impact declined and vanished. I continue taking NAD after that point and when I discontinued it there was no perceptible withdrawal affect. I reported on this in the latest blog entry, Part fourof the Wwearables series. This anecdotal result is consistent with the statements you quoted.
2. During this entire period, before after and now, I have also been supplementing daily with NR for several months now, I truly do not know what difference this makes, if any. When I run out of pills in about a week I will discontinue NR and see if it makes a difference in my heart rate stress biomarkers. So, in a month or so I should be in a better position to give a personal response to this part of your question. Vince
If the NAD boost from NR is transient because NQ01 will reestablish the baseline, will stopping NR drop NAD+ transiently until NQ01 reestablishes the norm?
That would mean that cycling, per Jon’s note above, would be of no benefit.
If there is a chance to update the “Supplements in Combined Firewalls” from 2008 that would be great. Thanks.
Jon
I would be also interested !
You do an awesome job.
Fabien.
Jon
Thank you thank for the reminder. I am painfully aware of how that document is becoming obsolete despite its many valuable features. I think I will updated it in two phases: a first update done soon to reflect my existing firewall regimen, probably with comments as to what is behind the changes. Then, a major rewrite probably requiring a month or two. There is generally nothing wrong with what is said in this treatise. However, several sections can be enhanced considerably by taking new or additional research into account. I am thinking of possibly making it into an e-book. Vince
2008 above should say instead 2012.
Thanks,
Jon
Vince, your articles are amazing. Thank you for putting in the time to do all this research. I was going to supplement with NAD+ (before I started reading your articles) to try to cure my autoimmune disease, which I know is affecting my kidneys and other organs. I have changed my diet probably 100 times, and nothing has worked. Doctors can’t figure out why I have autoimmune disease. So I guess I’m pressed to ask you for your recommendation for reversing aging, disease, inflammation with supplements?
Why speak about beta lapachone since it was shown being lethal with dose as low as 6 doses of 9mg ??
Pingback: MECHANISMS OF AGING AND INTERVENTIONS FOR LONGEVITY PART 1 THE FIRST 100 YEARS - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™