
By Vince Giuliano with inputs from James P. Watson
Accelerated by the publication in December 2013 of a seminal paper by David Sinclair and his US and Australian colleagues(ref), there has been increasing interest and excitement about the prospects of discovering means for offsetting age–related declines in NAD+ as a strategy for disease prevention and life extension. Actually, similar proposals have appeared in the literature for several years. However, for lay people and even many scientists, the scientific discussions of NAD+ are so complicated that they seem to be virtually unfathomable. In fact, understanding NAD+ and its roles requires fathoming a large number of related actors and their roles – complex metabolic pathways, an alphabet soup collection of enzymes and gene activation cofactors, the family of Sirtuins, biological processes in different cell compartments and organs, and multiple pathological and health-producing concomitants. The complex of related actors has come to be called the NAD World. It is not surprising that many intelligent people are left confused about it.
We have already referred to NAD+ and its health concomitants several times in this blog. In fact, Jim Watson characterized it as the most important scientific topic he encountered in 2013 and you might want to start out by reviewing what he said about it in this blog entry.
The purpose of this current Part 1 blog entry is to provide an overview treatment of the NAD World and its nuances: to identify the major molecular entities involved, their roles, health and longevity ramifications, the reasons for the current excitement, and to begin to clarify what is actually known and what the remaining uncertainties are. We also discuss a few important facets of the general topic which are ignored in many of the current publications, such as the high degree of circadian regulation of NAD+ .and about proteins that regulate NAD+ and its cousins such as CD38 and CCR2.
There will be additional Parts in this series. Part 2 concentrates on the reasons for focusing on NAD+, particularly with respect to interventions that are seriously likely to lead to longer healthier lives. Written mainly by Jim Watson, it discusses molecular processes in the NAD salvage cycle that are responsible for the health-inducing and life-extending properties of calorie restriction and further discuss the key roles of Sirtuins, SIRT1, SIRT6 and SIRT7 in particular. It goes into more detail in several areas covered less- deeply in the Part 1 blog entry. It points to some difficult-to-integrate contrarian research and basic uncertainties. Also, Jim and I are centrally concerned with developing a set of biomarkers that can tell us what the various approaches to enhancing body levels of NAD+ actually accomplish or fail to accomplish. We identify several candidate biomarkers in Part 2. A Part 3 blog entry in this NAD World series describes 30 Major Factors that Control SIRT1 expression, SIRT1 Activity, and SIRT1-mediated Aging. The Part 3 blog entry identifies 30 Major Factors that Control SIRT1 Expression, SIRT1 Activity, and SIRT1-mediated Aging. Also, we discuss the NAD+/NADH ratio and what affects it and the NQ01 gene. The Part 4 blog entry is concerned further with the NQ01 gene, the Warburg effect, SIRT 1 and inflammation, and possible interventions. The Part 5 blog entry in this NAD World series is concerned with the conflicting roles of extra-cellular NAMPT, which behaves as both an enzyme and a cytokine, and how it is implicated in inducing many disease processes as well as having some positive health impacts.
These blog entries lay out reasons why simply enhancing NAD+ levels, say by oral consumption of nicotinamide riboside, might or might not work to increase health or longevity. Nonetheless, my perception is that a big new area of longevity science is opening up, one that could offer new practical interventions that lead to longer healthier lives.
INTRODUCTIONS TO THE MAIN ACTORS AND THEIR RELATIONSHIPS — THE NAD WORLD
It was suggested back in 2009 that NAD and the molecules and pathways it dances with constitute a world for explaining metabolism and aging.
The 2009 publication The NAD world: A new systemic regulatory network for metabolism and aging-Sirt1, systemic NAD biosynthesis, and their importance related: “For the past several years, it has been demonstrated that the NAD-dependent protein deacetylase Sirt1 and nicotinamide phosphoribosyltransferase (Nampt)-mediated systemic NAD biosynthesis together play a critical role in the regulation of metabolism and possibly aging in mammals. Based on our recent studies on these two critical components, we have developed a hypothesis of a novel systemic regulatory network, named “NAD World”, for mammalian aging. Conceptually, in the NAD World, systemic NAD biosynthesis mediated by intra- and extracellular NAMPT functions as a driver that keeps up the pace of metabolism in multiple tissues/organs, and the NAD-dependent deacetylase Sirt1 serves as a universal mediator that executes metabolic effects in a tissue-dependent manner in response to changes in systemic NAD biosynthesis. This new concept of the NAD World provides important insights into a systemic regulatory mechanism that fundamentally connects metabolism and aging and also conveys the ideas of functional hierarchy and frailty for the regulation of metabolic robustness and aging in mammals.”
Here is a handy reference guide to some of the main molecular actors that will be showing up in the following discussions and their significance – a cast of main characters in the NAD World but by no means all of them.
NAD – Nicotinamide adenine dinucleotide (NAD) is a coenzyme found in all living cells. The compound is a dinucleotide, since it consists of two nucleotides joined through their phosphate groups. One nucleotide contains an adenine base and the other nicotinamide. Nicotinamide adenine dinucleotide exists in two forms, an oxidized and reduced form abbreviated as NAD+ and NADH respectively.(ref) As we will see, it is a molecule of central importance to metabolic and other key biological processes in humans.
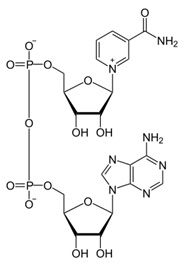
NAD+ – oxidized form of NAD, very important as a cofactor for health and longevity-related processes including DNA repair and mitochondrial operability as will be discussed. The central actor in our current drama, it acts as a biological oxidizing agent. Declines sharply with age negatively impacting on DNA repair and mitochondrial health and benefits conveyed by sirtuins.
NADH – reduced form of NAD. Acts as a biological reducing agent. Cycled back in the body to NAD+ by what are known as the NAD salvage pathways described below.
NA – Nicotinic acid (aka Niacin aka Vitamin B3) – Niacin –” is an organic compound with the formula C6H5NO2 and, depending on the definition used, one of the 20 to 80essential human nutrients.(ref)” Important in the current drama as a precursor to Nicotinamide which is a precursor to NAD. Unfortunately, consuming niacin is generally not a good way to enhance body levels of NAD since its metabolite niacinamide inhibits the sirtuins.
NAM – Nicotinamide (also known as niacinamide and nicotinic amide), “is the amide of nicotinic acid (vitamin B3 / niacin). Nicotinamide is a water-soluble vitamin and is part of the vitamin B group. Nicotinic acid, also known as niacin, is converted to nicotinamide in vivo.”(ref). It is a precursor of NAD but there is a catch to simply supplementing with it. It inhibits the sirtuins and their health-producing properties.
NR – Nicotinamide riboside – a precursor of NAD, and is a source of Vitamin B3. Available as a commercial dietary supplement thought to promote NAD+. One of the possible approaches to enhancing body levels of NAD+(ref)
NAD Salvage Pathways – “Besides assembling NAD+ de novo from simple amino acid precursors, cells also salvage preformed compounds containing nicotinamide. Although other precursors are known, the three natural compounds containing the nicotinamide ring and used in these salvage metabolic pathways are nicotinic acid (Na), nicotinamide (Nam) and nicotinamide riboside (NR).[2] These compounds can be taken up from the diet, where the mixture of nicotinic acid and nicotinamide are called vitamin B3 or niacin. However, these compounds are also produced within cells, when the nicotinamide moiety is released from NAD+ in ADP-ribose transfer reactions. Indeed, the enzymes involved in these salvage pathways appear to be concentrated in the cell nucleus, which may compensate for the high level of reactions that consume NAD+ in this organelle.[26] Cells can also take up extracellular NAD+ from their surroundings[27](ref).”
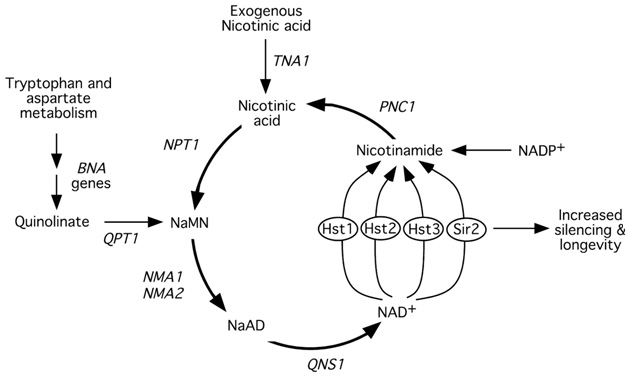
Image and legend source is the 2002 publication Manipulation of a Nuclear NAD+ Salvage Pathway Delays Aging without Altering Steady-state NAD+ Levels: ” Model for life span extension via increased flux through the NAD+ salvage pathway. – Type III histone deacetylases such as Sir2 and Hst1–4 catalyze a key step in the salvage pathway by converting NAD+ to nicotinamide. Additional copies of PNC1, NPT1, NMA1, and NMA2 increase flux through the NAD+salvage pathway, which stimulates Sir2 activity and increases life span. Additional copies of QNS1 fail to increase silencing. Unlike other steps in the pathway, its substrate cannot be supplied from a source outside the salvage pathway and is therefore limiting for the reaction. Abbreviations: NAD +, nicotinamide adenine dinucleotide; NaMN, nicotinic acid mononucleotide; NaAD, desamido-NAD+(ref).” A number of important matters are not shown in this simplified diagram, such as ATP (required as an energy source in the lower loop to make NAD), the key roles of SIRT1, and CD38 which eats up NAD+.
NADPH – is the reduced form of NADP+. “NADPH provides the reducing equivalents for biosynthetic reactions and the oxidation-reduction involved in protecting against the toxicity of ROS (reactive oxygen species), allowing the regeneration of GSH (reduced glutathione).[3] NADPH is also used for anabolic pathways, such as lipid synthesis, cholesterol synthesis, and fatty acid chain elongation. — The NADPH system is also responsible for generating free radicals in immune cells. These radicals are used to destroy pathogens in a process termed the respiratory burst.[4] It is the source of reducing equivalents for cytochrome P450 hydroxylation of aromatic compounds, steroids, alcohols, and drugs(ref).”
NAADP – Nicotinic acid adenine dinucleotide phosphate “(NAADP) is one of the most potent stimulators of intracellular Ca2+ release known to date. The role of the NAADP system in physiological processes is being extensively investigated at the present time. Exciting new discoveries in the last 5 years suggest that the NAADP-regulated system may have a significant role in intracellular Ca2+ signaling. The NAADP receptor and its associated Ca2+ pool have been hypothesized to be important in several physiological processes including fertilization, T cell activation, and pancreatic secretion(ref).”
NADPH oxidase – “Nicotinamide adenine dinucleotide phosphate-oxidase is a membrane-bound enzyme complex. It can be found in the plasma membrane as well as in the membranes of phagosomes used by neutrophil white blood cells to engulf microorganisms. — NADPH oxidase generates superoxide by transferring electrons from NADPH inside the cell across the membrane and coupling these to molecular oxygen to produce superoxide anion, a reactive free-radical. Superoxide can be produced in phagosomes, which contain ingested bacteria and fungi, or it can be produced outside of the cell. In a phagosome, superoxide can spontaneously form hydrogen peroxide that will undergo further reactions to generate reactive oxygen species (ROS)(ref).” Plays important roles in generating ROS to deal with pathogens.
NAMPT – “Nicotinamide phosphoribosyltransferase (NAmPRTase or NAMPT) also known as pre-B-cell colony-enhancing factor 1 (PBEF1) or visfatin is an enzyme that in humans is encoded by the PBEF1 gene.[1] This protein has also been reported to be a cytokine (PBEF) that promotes B cell maturation and inhibits neutrophil apoptosis. — This enzyme participates in nicotinate and nicotinamide metabolism.(ref)” NAMPT is the rate-limiting enzyme of the NAD(+) salvage pathway and enhances SIRT1 activity by increasing the amount of NAD+.
NMNAT – Nicotinamide/nicotinic acid mononucleotide adenylyltransferase An enzyme that plays a role in NAD synthesis. “– a rate-limiting enzyme present in all organisms, reversibly catalyzes the important step in the biosynthesis of NAD from ATP and NMN. NAD and NADP are used reversibly in anabolic and catabolic reactions(ref).”
SIRTUINS (Silent Information Regulators SIRT1 – SIRT7) Sirtuins extend the lifespans of lower ife forms including yeast, nematodes and drosphila, and appear to have pluripotent health effects in humans. “Sirtuin or Sir2 proteins are a class of proteins that possess either mono-ADP-ribosyltransferase, or deacylase activity, including deacetylase, desuccinylase, demalonylase, demyristoylase and depalmitoylase activity.[2][3][4][5] Sirtuins regulate important biological pathways in bacteria, archaea and eukaryotes. The name Sir2 comes from the yeast gene ‘silent mating-type information regulation 2’,[6] the gene responsible for cellular regulation in yeast. Sirtuins have been implicated in influencing a wide range of cellular processes like aging, transcription, apoptosis, inflammation[7] and stress resistance, as well as energy efficiency and alertness during low-calorie situations.[8] Sirtuins can also control circadian clocks and mitochondrial biogenesis(ref).” Sirtuins convert NAD(+) into nicotinamide (NAM). The sirtuins vary in their functions and in general serve as deacetylases. We have written about them a number of times in this blog(ref), and we will comment further here on some of their specific functions. SIRT1, in particular, the mammalian counterpart of SIR2, both depends on the presence of NAD+ for its activation and has important regulatory functions in the NAD salvage pathway. “SIRT1 is required to regulate adaptive responses to acute and chronic energy limitations, such as fasting and dietary restriction. In general, the evolutionary role of SIR2 ones is thought to be that they provide mechanisms through which an organism can adopt to changes in environment and diet. An example of the possible role of SIRT6 could be to allow animals to switch between vegetarian and meat diets. Sirtuins are completely dependent on the availability of NAD+ for their formation and thus are central actor’s in the NAD World. An important negative consequence of insufficient levels of NAD+ is insufficient levels of sirtuins and compromise of their important biological activities.” A table showing the deacetylase and deacelase activities of the seven sirtuin sisters can be found on this site.
CCAR2 (Cell Cycle And Apoptosis Regulator 2, also known as DBC1) An important negative regulator of SIRT1 (ref) and a consumer of NAD+.
CD38 – “CD38 (cluster of differentiation 38), also known as cyclic ADP ribose hydrolase is a glycoprotein[1] found on the surface of many immune cells (white blood cells), including CD4+, CD8+, B lymphocytes and natural killer cells. — CD38 is a multifunctional ectoenzyme that catalyzes the synthesis and hydrolysis of cyclic ADP-ribose (cADPR) from NAD+ to ADP-ribose. These reaction products are essential for the regulation of intracellular Ca2+[5] (ref).” CD38 can be a major consumer of NAD as a substrate.
MNA – Methylnicotinamide – ” Methylnicotinamide is a metabolite of nicotinamide and is produced primarily in the liver. It has anti-inflammatory properties (PMID 16197374 ). It is a product of nicotinamide N-methyltransferase [EC 2.1.1.1] in the pathway of nicotinate and nicotinamide metabolism (KEGG). 1-Methylnicotinamide may be an endogenous activator of prostacyclin production and thus may regulate thrombotic as well as inflammatory processes in the cardiovascular system(ref).”
PARP – “Poly (ADP-ribose) polymerase (PARP) is a family of proteins involved in a number of cellular processes involving mainly DNA repair and programmed cell death. — The PARP family comprises 17 members (10 putative). They have all very different structures and functions in the cell.– PARP is found in the cell’s nucleus. The main role is to detect and signal single-strand DNA breaks (SSB) to the enzymatic machinery involved in the SSB repair. PARP activation is an immediate cellular response to metabolic, chemical, or radiation-induced DNA SSB damage. Once PARP detects a SSB, it binds to the DNA, and, after a structural change, begins the synthesis of a poly (ADP-ribose) chain (PAR) as a signal for the other DNA-repairing enzymes such as DNA ligase III (LigIII), DNA polymerase beta (polβ), and scaffolding proteins such as X-ray cross-complementing gene 1 (XRCC1). After repairing, the PAR chains are degraded via Poly(ADP-ribose) glycohydrolase(PARG).[1] — It is interesting to note that NAD+ is required as substrate for generating ADP-ribose monomers. The overactivation of PARP may deplete the stores of cellular NAD+ and induce a progressive ATP depletion and necrotic cell death, since glucose oxidation is inhibited. In this regard, PARP is inactivated by caspase-3 cleavage (in a specific domain of the enzyme) during programmed cell death. — PARP enzymes are essential in a number of cellular functions,[2] including expression of inflammatory genes:[3] PARP1 is required for the induction of ICAM-1 gene expression by smooth muscle cells, in response to TNF[4] (ref).”
ATP – ” Adenosine triphosphate (ATP) is a nucleoside triphosphate used in cells as a coenzyme, often called the “molecular unit of currency” of intracellular energy transfer.[1] — ATP transports chemical energy within cells for metabolism(ref).”
ADP – Adenosine diphosphate, abbreviated ADP, is an important organic compound in metabolism and is essential to the flow of energy in living cells. — The cleavage of a phosphate group from ATP results in the coupling of energy to metabolic reactions and a by-product, a molecule of ADP.[1] Being the “molecular unit of currency”, ATP is continually being formed from lower-energy molecules of ADP and AMP(ref).”.
AMP – Adenosine monophosphate, NAD can be synthesized from it(ref),
“AMP, ADP, ATP – ATP consists of three phosphate groups attached in series to the 5’ carbon location, whereas ADP contains two phosphate groups attached to the 5’ position, and AMP contains only one phosphate group attached at the 5’ position. Energy transfer used by all living things is a result of dephosphorylation of ATP by enzymes known as ATPases. The cleavage of a phosphate group from ATP results in the coupling of energy to metabolic reactions and a by-product, a molecule of ADP.[1] Being the “molecular unit of currency”, ATP is continually being formed from lower-energy molecules of ADP and AMP. The biosynthesis of ATP is achieved throughout processes such as substrate-level phosphorylation, oxidative phosphorylation, and photophosphorylation, all of which facilitating the addition of a phosphate group to an ADP molecule.”(ref)
AMPK – ” AMP-activated protein kinase (AMPK) plays a key role as a master regulator of cellular energy homeostasis. The kinase is activated in response to stresses that deplete cellular ATP supplies such as low glucose, hypoxia, ischemia, and heat shock(ref).”
cAMP – “AMP can also exist as a cyclic structure known as cyclic AMP (or cAMP). Within certain cells the enzyme adenylate cyclase makes cAMP from ATP, and typically this reaction is regulated by hormones such as adrenaline or glucagon. cAMP plays an important role in intracellular signaling. (ref)
CREB – cAMP response element-binding protein[1] – is a cellular transcription factor. It binds to certain DNA sequences calledcAMP response elements (CRE), thereby increasing or decreasing the transcription of the downstream genes.[2] CREB was first described in 1987 as a cAMP-responsive transcription factor regulating the somatostatin gene[3] (ref)”
PGC1 alpha – “PGC-1-alpha (Peroxisome proliferator-activated receptor gamma coactivator 1-alpha) “is a protein that in humans is encoded by thePPARGC1A gene.[1] The protein encoded by this gene is a transcriptional coactivator that regulates the genes involved in energy metabolism. This protein interacts with the nuclear receptor PPAR-gamma, which permits the interaction of this protein with multiple transcription factors. This protein can interact with, and regulate the activities of, cAMP response element binding protein (CREB) and nuclear respiratory factors (NRFs). It provides a direct link between external physiological stimuli and the regulation of mitochondrial biogenesis, and is a major factor that regulates muscle fiber type determination. This protein may be also involved in controlling blood pressure, regulating cellular cholesterol homoeostasis, and the development of obesity[2] (ref).” We have discussed PGC-1-alpha several times in this blog (ref).
TOP LEVEL POINTS
My approach in this blog entry will be top-down, starting with general points that establish the importance of the topic to health and longevity – and finishing with some of the more-technical detail that conveys so much richness to the topic. Here is a list of top-level points that will be detailed in the following citations:
- NAD levels decline precipitously with aging in mammals and humans, and are strongly negatively affected by multiple disease and stress processes including obesity. At advanced ages these levels are a tiny fraction of what they are in young people.
- Maintaining a high level of constitutive NAD+ in the body is critical for maintaining health and functionality, for averting or reversing a number of deleterious disease phenomena, for energy and vitality, and possibly for extending life spans in humans.
- NAD+ is a key substrate for the production on SIRT1 and other sirtuins needed for proper histone deacetylation and gene regulation in response to changing conditions. Its availability is also critical for preventing a state of pseudohypoxia in cell nuclii and proper generation of mitochondrial proteins required for efficient electron transfer chain operation.
- Inadequate levels of NAD can negatively impact on DNA repair, runaway inflammation, normal metabolism, responses to oxidative stress, mitochondrial health and electron transfer chain efficiency in mitichondria, and lead to the Warburg Effect of pathological metabolism.
- NAD+ is needed for responsiveness to oxidative stresses and for control of inflammatory processes
- NAD+ deficiency thus impairs a. Proper DNA damage repair, b. Production of sirtuins and processes that depend on sirtuins for homeostasis and health, c. Control of gene activation, d. Mitochondrial functionality and viability, e. Capacity to deal with injuries and pathological situations, and f. Ability to control excess inflammation.
- The rate of DNA damage increases significantly with aging, increasing demand for NAD+ for DNA repair. However in general, the reasons for age-related decline in NAD are not well understood.
- As a consequence of the above, inadequate levels of NAD can lead to a laundry list of disease processes, including type 2 diabetes, Alzheimer’s disease, and cancer. And to acceleration of aging. Indeed, I venture an opinion that there is an unvirtuous cyclic process of interaction of the NAD-related feedback loops that manifests itself in the progressive acceleration of the processes of aging with aging itself – why we generally age faster and faster as we get older.
- There is a great deal of research knowledge about what generates NAD, what runs it down, its relationship to metabolism and other metabolic factors, the biological processes affecting it or affected by it, and how it’s expression relates to multiple disease processes.
- It has long been known that many deleterious disease phenomena can be prevented or reversed by promoting higher levels of NAD in animal models. There appears to be ample evidence that this approach is generally safe.
- For a number of years it has been proposed in research publications that promoting NAD levels in humans could be an effective preventative and/or therapeutic strategy.for multiple disease processes Also, it is a proven strategy for extending the lifespans of c-elegans worms. However, this strategy has never entered our mainline medical or health maintenance system.
- Multiple strategies have been investigated for enhancing human levels of NAD, including enhancing its original synthesis in the body, downregulating biological processes that consume it, ingesting precursor molecules such as NMN or NR, and direct introduction of NAD into the bloodstream via IV. At present, we do not know which of these approaches will be most efficacious and cost-effective.
I will amplify on these points by citing selections from many relevant publications that describe aspects of the NAD World. After this, I go on to second-level of selected key points that relate to the biological mechanisms involved – for example, the key systematic roles that NAD plays in regulating metabolism and aging, and how NAD is consumed as an important substrate for DNA repair processes. Topics 11 and 12 above will mainly be discussed in further blog entries in this series.
RESEARCH RESULTS
The 2013 publication The NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling reports: “NAD+ is an important co-factor regulating metabolic homeostasis and a rate-limiting substrate for sirtuin deacylase. We show that NAD+ levels are reduced in aged mice and C. elegans and that decreasing NAD+ levels results in a further reduction in worm lifespan. Conversely, genetic or pharmacological restoration of NAD+ prevents age-associated metabolic decline and promotes longevity in worms. These effects are dependent upon the protein deacetylase sir-2.1 and involve the induction of mitonuclear protein imbalance as well as activation of stress signaling via the mitochondrial unfolded protein response (UPRmt) and the nuclear translocation and activation of FOXO transcription factor DAF-16. Our data suggest that augmenting mitochondrial stress signaling through the modulation of NAD+ levels may be a target to improve mitochondrial function and prevent or treat age-associated decline. — Alterations in NAD+ levels have a powerful metabolic impact, since it serves as an obligatory substrate for the deacetylase activity of the sirtuin proteins (Guarente, 2008; Haigis and Sinclair, 2010; Houtkooper et al., 2010a). The best-characterized mammalian sirtuin is SIRT1, which controls mitochondrial function through the deacetylation of targets that include PGC-1α and FOXO (Chalkiadaki and Guarente, 2012; Houtkooper et al., 2012). The administration of NAD+ precursors, such as nicotinamide mononucleotide (Yoshino et al., 2011) or nicotinamide riboside (NR) (Canto et al., 2012), has proven to be an efficient way to increase NAD+ levels and SIRT1 activity, improving metabolic homeostasis in mice. Furthermore, the NAD+-consuming poly(ADP-ribose) polymerase proteins—with PARP1 and PARP2 representing the main PARP activities in mammals—were classically described as DNA repair proteins (Gibson and Kraus, 2012; Schreiber et al., 2006), but recent studies have linked these proteins to metabolism (Asher et al., 2010; Bai et al., 2011a; Bai et al., 2011b; Erener et al., 2012). Indeed, genetic or pharmacological inactivation of PARP1 increased tissue NAD+ levels and activated mitochondrial metabolism (Bai et al., 2011b). An association between PARPs and lifespan has been postulated (Grube and Burkle, 1992; Mangerich et al., 2010), but a causal role remained unclear. A final line of evidence in support of a role for NAD+ in metabolic control came from the deletion of an alternative NAD+-consuming protein, CD38, which also led to NAD+ accumulation and subsequent SIRT1 activation in mice, and proved protective against high-fat diet-induced obesity (Barbosa et al., 2007). — Considering the intimate link between metabolism and longevity (Guarente, 2008; Houtkooper et al., 2010b), we hypothesized that increasing NAD+ levels may be sufficient to increase mitochondrial activity and extend lifespan (Houtkooper and Auwerx, 2012). Here we show how supplementation of PARP inhibitors or NAD+ precursors led to improved mitochondrial homeostasis through the activation of the worm sirtuin homolog, sir-2.1. ”
Because the NAD World pathways are evolutionarily conserved in mammals and humans, there is reason to believe these mechanisms apply to us humans as well.
The 2013 publication The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing reports: “Ageing is associated with a variety of pathophysiological changes, including development of insulin resistance, progressive decline in β-cell function and chronic inflammation, all of which affect metabolic homeostasis in response to nutritional and environmental stimuli. SIRT1, the mammalian nicotinamide adenine dinucleotide (NAD)-dependent protein deacetylase, and nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting NAD biosynthetic enzyme, together comprise a novel systemic regulatory network, named the ‘NAD World’, that orchestrates physiological responses to internal and external perturbations and maintains the robustness of the physiological system in mammals. In the past decade, an accumulating body of evidence has demonstrated that SIRT1 and NAMPT, two essential components in the NAD World, play a critical role in regulating insulin sensitivity and insulin secretion throughout the body. In this article, we will summarize the physiological significance of SIRT1 and NAMPT-mediated NAD biosynthesis in metabolic regulation and discuss the ideas of functional hierarchy and frailty in determining the robustness of the system. We will also discuss the potential of key NAD intermediates as effective nutriceuticals for the prevention and the treatment of age-associated metabolic complications, such as type 2 diabetes.”
As mentioned above, sirtuins depend on the availability of NAD+ as a substrate and some of the negative consequences of insufficient NAD+ are associated with surtuin insufficiency. Likewise, there is evidence that high expression of sirtuins associated with high levels of NAD+, SIRT1 and the others as well, convey a variety of longevity benefits.
One way this shows up is slowing or averting stem cell senescence. The 2014 publication SIRT1 ameliorates age-related senescence of mesenchymal stem cells via modulating telomere shelterin reports: “Mesenchymal stem cells (MSCs) senescence is an age-related process that impairs the capacity for tissue repair and compromises the clinical use of autologous MSCs for tissue regeneration. Here, we describe the effects of SIRT1, a NAD(+)-dependent deacetylase, on age-related MSCs senescence. Knockdown of SIRT1 in young MSCs induced cellular senescence and inhibited cell proliferation whereas overexpression of SIRT1 in aged MSCs reversed the senescence phenotype and stimulated cell proliferation. These results suggest that SIRT1 plays a key role in modulating age-induced MSCs senescence. Aging-related proteins, P16 and P21 may be downstream effectors of the SIRT1-mediated anti-aging effects. SIRT1 protected MSCs from age-related DNA damage, induced telomerase reverse transcriptase (TERT) expression and enhanced telomerase activity but did not affect telomere length. SIRT1 positively regulated the expression of tripeptidyl peptidase 1 (TPP1), a component of the shelterin pathway that protects chromosome ends from DNA damage. Together, the results demonstrate that SIRT1 quenches age-related MSCs senescence by mechanisms that include enhanced TPP1 expression, increased telomerase activity and reduced DNA damage.”
The 2011 publication Dissecting systemic control of metabolism and aging in the NAD World relates “Accumulating bodies of evidence have suggested that SIRT1 plays an important role in retarding age-associated pathophysiological changes and preventing from diseases of aging, such as type 2 diabetes, Alzheimer’s disease, and cancer. Whole-body SIRT1-overexpressing transgenic mice show significant protection from the adverse effects of high-fat diet or aging on glucose metabolism [9] and [10]. SIRT1-activating compounds are also able to improve glucose homeostasis and insulin sensitivity in diet-induced and genetic type 2 diabetes animal models [9] and [10]. ”
From the same publication: “It has recently been demonstrated that SIRT1 prevents two critical pathological aspects of Alzheimer’s disease: Aβ amyloid deposition and tauopathy. SIRT1 decreases the production of Aβ amyloid by deacetylating the retinoic acid receptor β and thereby up-regulating ADAM10, a major component of α-secretase [19]. SIRT1 also promotes degradation of phosphorylated tau by deacetylating it and prevents tau-mediated neurodegeneration [20]. Furthermore, SIRT1 regulates memory and synaptic plasticity, providing insight into potential intervention against age-associated cognitive disorders [21] and [22]. It has also been reported that SIRT1 transgenic mice show a lower incidence of spontaneous carcinomas and sarcomas and a reduced susceptibility to high-fat diet/carcinogen-induced liver tumors, compared to wild-type control mice [23]. These findings provide strong support for the importance of SIRT1 in the prevention of major age-associated diseases.”
The 2011 publication NAD+ treatment decreases tumor cell survival by inducing oxidative stress reports: “NAD+ plays important roles in various biological processes. It has been shown that NAD+ treatment can decrease genotoxic agent-induced death of primary neuronal and astrocyte cultures, and NAD+ administration can reduce ischemic brain damage. However, the effects of NAD+ treatment on tumor cell survival are unknown. In this study we found that treatment of NAD+ at concentrations from 10 micromolar to 1 mM can significantly decrease the survival of various types of tumor cells such as C6 glioma cells. In contrast, NAD+ treatment did not impair the survival of primary astrocyte cultures. Our study has also indicated that oxidative stress mediates the effects of NAD+ on the survival of tumor cells, and P2X7 receptors and altered calcium homeostasis are involved in the effects of NAD+ on the cell survival. Collectively, our study has provided the first evidence that NAD+ treatment can decrease the survival of tumor cells by such mechanisms as inducing oxidative stress. Because NAD+ treatment can selectively decrease the survival of tumor cells, NAD+ may become a novel agent for treating cancer.”
The 2012 publication Age-associated changes in oxidative stress and NAD+ metabolism in human tissue reports: “Nicotinamide adenine dinucleotide (NAD(+)) is an essential electron transporter in mitochondrial respiration and oxidative phosphorylation. In genomic DNA, NAD(+) also represents the sole substrate for the nuclear repair enzyme, poly(ADP-ribose) polymerase (PARP) and the sirtuin family of NAD-dependent histone deacetylases. Age associated increases in oxidative nuclear damage have been associated with PARP-mediated NAD(+) depletion and loss of SIRT1 activity in rodents. In this study, we further investigated whether these same associations were present in aging human tissue. Human pelvic skin samples were obtained from consenting patients aged between 15-77 and newborn babies (0-1 year old) (n = 49) previously scheduled for an unrelated surgical procedure. DNA damage correlated strongly with age in both males (p = 0.029; r = 0.490) and females (p = 0.003; r = 0.600) whereas lipid oxidation (MDA) levels increased with age in males (p = 0.004; r = 0.623) but not females (p = 0.3734; r = 0.200). PARP activity significantly increased with age in males (p<0.0001; r = 0.768) and inversely correlated with tissue NAD(+) levels (p = 0.0003; r = -0.639). These associations were less evident in females. A strong negative correlation was observed between NAD(+) levels and age in both males (p = 0.001; r = -0.706) and females (p = 0.01; r = -0.537). SIRT1 activity also negatively correlated with age in males (p = 0.007; r = -0.612) but not in females. Strong positive correlations were also observed between lipid peroxidation and DNA damage (p<0.0001; r = 0.4962), and PARP activity and NAD(+) levels (p = 0.0213; r = 0.5241) in post pubescent males. This study provides quantitative evidence in support of the hypothesis that hyperactivation of PARP due to an accumulation of oxidative damage to DNA during aging may be responsible for increased NAD(+) catabolism in human tissue. The resulting NAD(+) depletion may play a major role in the aging process, by limiting energy production, DNA repair and genomic signalling.”
What eats up and depletes NAD?
In the normal operation of the NAD salvage cycle, NAD is conserved, shuttled back and forth between its two forms NAD+ and NADH. In this process, NAD acts as a cofactor in a major metabolic cycle. In other vital processes, however, NAD acts as a substrate and is consumed. It is quite possibly the case that age-related decline in levels of NAD are mainly associated with increased demand by those substrate processes with aging. Here I discuss consumption of NAD by 1. PARPS, 2. Production of sirtuins, and 3. CD38 I also mention 4. CCAR2, a negative regulator of SIRT1 which can nullify health benefits of the availability of NAD+.
1. PARPS are major consumers of NAD+, using it for DNA repair.
Probably they are the largest consumer of NAD+, especially in older people where the need for DNA repair accelerates with age. As a matter of fact, one strategy for increasing levels of NAD in mice is to inhibit PARPs, Although this strategy seems to increase lifespans of mice by making more NAD available to mitochondria, I suspect it is a shortsighted approach when it comes to human health and longevity, ” Nuclear poly(ADP ribose) polymerase 1 (1) generates polyADP ribose and probably accounts for the majority of nuclear NAD+ degradation. Direct interaction with NMNAT1 facilitates NAD+ supply to activated PARP1.(ref)”
The 2011 publication PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation reports: “SIRT1 regulates energy homeostasis by controlling the acetylation status and activity of a number of enzymes and transcriptional regulators. The fact that NAD(+) levels control SIRT1 activity confers a hypothetical basis for the design of new strategies to activate SIRT1 by increasing NAD(+) availability. Here we show that the deletion of the poly(ADP-ribose) polymerase-1 (PARP-1) gene, encoding a major NAD(+)-consuming enzyme, increases NAD(+) content and SIRT1 activity in brown adipose tissue and muscle. PARP-1(-/-) mice phenocopied many aspects of SIRT1 activation, such as a higher mitochondrial content, increased energy expenditure, and protection against metabolic disease. Also, the pharmacologic inhibition of PARP in vitro and in vivo increased NAD(+) content and SIRT1 activity and enhanced oxidative metabolism. These data show how PARP-1 inhibition has strong metabolic implications through the modulation of SIRT1 activity, a property that could be useful in the management not only of metabolic diseases, but also of cancer.”
2. Sirtuins also draw down on NAD+ as a substrate, although the exact mechanisms of sirtuin activation are not clear to me.
From the 2011 publication Mammalian Sirtuins and Energy Metabolism “Despite diverse subcellular localizations and a broad range of substrate specificities, the activity of all sirtuins is directly controlled by cellular NAD+ levels, which is an indicator of cellular metabolic status. The activity of these enzymes is also inhibited by their common enzymatic product, nicotinamide (19), and possibly by NADH (20). It is therefore not surprising that the activity of sirtuins changes in response to environmental cues that impact cellular metabolic state.”
The 2011 publication Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats reports: “The cofactor nicotinamide adenine dinucleotide (NAD+) has emerged as a key regulator of metabolism, stress resistance and longevity. Apart from its role as an important redox carrier, NAD+ also serves as the sole substrate for NAD-dependent enzymes, including poly(ADP-ribose) polymerase (PARP), an important DNA nick sensor, and NAD-dependent histone deacetylases, Sirtuins which play an important role in a wide variety of processes, including senescence, apoptosis, differentiation, and aging. We examined the effect of aging on intracellular NAD+ metabolism in the whole heart, lung, liver and kidney of female wistar rats. Our results are the first to show a significant decline in intracellular NAD+ levels and NAD:NADH ratio in all organs by middle age (i.e.12 months) compared to young (i.e. 3 month old) rats. These changes in [NAD(H)] occurred in parallel with an increase in lipid peroxidation and protein carbonyls (o- and m- tyrosine) formation and decline in total antioxidant capacity in these organs. An age dependent increase in DNA damage (phosphorylated H2AX) was also observed in these same organs. Decreased Sirt1 activity and increased acetylated p53 were observed in organ tissues in parallel with the drop in NAD+ and moderate over-expression of Sirt1 protein. Reduced mitochondrial activity of complex I-IV was also observed in aging animals, impacting both redox status and ATP production. The strong positive correlation observed between DNA damage associated NAD+ depletion and Sirt1 activity suggests that adequate NAD+ concentrations may be an important longevity assurance factor.“
The 2012 publication The NAD+-dependent protein deacetylase activity of SIRT1 is regulated by its oligomeric status reports “SIRT1, a NAD+-dependent protein deacetylase, is an important regulator in cellular stress response and energy metabolism. While the list of SIRT1 substrates is growing, how the activity of SIRT1 is regulated remains unclear. We have previously reported that SIRT1 is activated by phosphorylation at a conserved Thr522 residue in response to environmental stress. Here we demonstrate that phosphorylation of Thr522 activates SIRT1 through modulation of its oligomeric status. We provide evidence that nonphosphorylated SIRT1 protein is aggregation-prone in vitro and in cultured cells. Conversely, phosphorylated SIRT1 protein is largely in the monomeric state and more active. Our findings reveal a novel mechanism for environmental regulation of SIRT1 activity, which may have important implications in understanding the molecular mechanism of stress response, cell survival, and aging.”
3. CD38 is a key enzyme sitting on the outside of the cell surface that is a major consumer of NAD that plays an important role in cell calcium homeostasis
From Mouse embryonic fibroblasts from CD38 knockout mice are resistant to oxidative stresses through inhibition of reactive oxygen species production and Ca(2+) overload: “CD38 is a multifunctional enzyme that has both ADP-ribosyl cyclase and cADPR hydrolase activities, being capable of cleaving NAD(+) to cyclic ADP ribose (cADPR) and hydrolyzing cADPR to ADPR. It has been reported that there is markedly a reduction of cADPR and elevation of NAD in many tissues from CD38 knockout (CD38(-/-)) mice. Cyclic ADPR is a potent second messenger for intracellular Ca(2+) mobilization, and NAD is a key cellular metabolite for cellular energetic and a crucial regulator for multiple signaling pathways in cells. We hypothesize that CD38 knockout may have a protective effect in oxidative stresses through elevating NAD and decreasing cADPR. In the present study, we observed that the mouse embryonic fibroblasts (MEFs) from CD38(-/-) mice were significantly resistant to oxidative stress such as H(2)O(2) injury and hypoxia/reoxygenation compared with wild type MEFs (WT MEFs). We further found that production of reactive oxygen species (ROS) and concentrations of intracellular Ca(2+) ([Ca(2+)](i)) in CD38(-/-) MEFs were markedly reduced compared with WT MEFs during hypoxia/reoxygenation. Coincidence with these results, a remarkably lower mRNA level of Nox1, one of the enzymes responsible for ROS generation, was observed in CD38(-/-) MEFs. Furthermore, we found that transcription of Nox1 mRNA in WT MEFs could be elevated by calcium ionophore ionomycin in a dose-dependent manner, indicating that the expression of Nox1 mRNA can be regulated by elevation of intracellular [Ca(2+)]. Therefore we concluded that CD38(-/-) MEFs are resistant to oxidative stresses through inhibiting intracellular Ca(2+) overload and ROS production which may be regulated by Ca(2+)-mediated inhibition of Nox1 expression. Our data should provide an insight for elucidating the roles of CD38 in oxidative stresses and a novel perspective of dealing with the ischemia/reperfusion-related diseases.”

Image and legend source “CD38, a transmembrane glycoprotein with ADP-ribosyl cyclase activity, catalyses the formation of Ca2+ signalling molecules and triggers proliferation and immune responses in lymphocytes — These results indicate that CD38 has a key role in neuropeptide release, thereby critically regulating maternal and social behaviours, and may be an element in neurodevelopmental disorders.”
NAD+ can be pumped out of cells to make CD38
The 2011 publication Connexin-43 hemichannels mediate cyclic ADP-ribose generation and its Ca2+-mobilizing activity by NAD+/cyclic ADP-ribose transport reports: “The ADP-ribosyl cyclase CD38 whose catalytic domain resides in outside of the cell surface produces the second messenger cyclic ADP-ribose (cADPR) from NAD(+). cADPR increases intracellular Ca(2+) through the intracellular ryanodine receptor/Ca(2+) release channel (RyR). It has been known that intracellular NAD(+) approaches ecto-CD38 via its export by connexin (Cx43) hemichannels, a component of gap junctions. — Our data suggest that Cx43 has a dual function exporting NAD(+) and importing cADPR into the cell to activate intracellular calcium mobilization.”
CD38 and its NAD+ and SIRT1 reducing effects can be inhibited by the phytosubstances quercetin and apigenin
The 2013 pubication Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome reports: “Metabolic syndrome is a growing health problem worldwide. It is therefore imperative to develop new strategies to treat this pathology. In the past years, the manipulation of NAD(+) metabolism has emerged as a plausible strategy to ameliorate metabolic syndrome. In particular, an increase in cellular NAD(+) levels has beneficial effects, likely because of the activation of sirtuins. Previously, we reported that CD38 is the primary NAD(+)ase in mammals. Moreover, CD38 knockout mice have higher NAD(+) levels and are protected against obesity and metabolic syndrome. Here, we show that CD38 regulates global protein acetylation through changes in NAD(+) levels and sirtuin activity. In addition, we characterize two CD38 inhibitors: quercetin and apigenin. We show that pharmacological inhibition of CD38 results in higher intracellular NAD(+) levels and that treatment of cell cultures with apigenin decreases global acetylation as well as the acetylation of p53 and RelA-p65. Finally, apigenin administration to obese mice increases NAD(+) levels, decreases global protein acetylation, and improves several aspects of glucose and lipid homeostasis. Our results show that CD38 is a novel pharmacological target to treat metabolic diseases via NAD(+)-dependent pathways.”
4. CCAR2 can be an important negative regulator of SIRT1
CCAR2 was until very recently called DBC1 after what was originally thought to be its main property (deleted in breast cabcer), so most of the research literature relating to it talks only about DBC1.
The 2012 publication Role of deleted in breast cancer 1 (DBC1) protein in SIRT1 deacetylase activation induced by protein kinase A and AMP-activated protein kinase reports: “The NAD(+)-dependent deacetylase SIRT1 is a key regulator of several aspects of metabolism and aging. SIRT1 activation is beneficial for several human diseases, including metabolic syndrome, diabetes, obesity, liver steatosis, and Alzheimer disease. We have recently shown that the protein deleted in breast cancer 1 (DBC1) is a key regulator of SIRT1 activity in vivo. Furthermore, SIRT1 and DBC1 form a dynamic complex that is regulated by the energetic state of the organism. Understanding how the interaction between SIRT1 and DBC1 is regulated is therefore essential to design strategies aimed to activate SIRT1. Here, we investigated which pathways can lead to the dissociation of SIRT1 and DBC1 and consequently to SIRT1 activation. We observed that PKA activation leads to a fast and transient activation of SIRT1 that isDBC1-dependent. In fact, an increase in cAMP/PKA activity resulted in the dissociation of SIRT1 and DBC1 in an AMP-activated protein kinase (AMPK)-dependent manner. Pharmacological AMPK activation led to SIRT1 activation by a DBC1-dependent mechanism. Indeed, we found that AMPK activators promote SIRT1-DBC1 dissociation in cells, resulting in an increase in SIRT1 activity. In addition, we observed that the SIRT1 activation promoted by PKA and AMPK occurs without changes in the intracellular levels of NAD(+). We propose that PKA and AMPK can acutely activate SIRT1 by inducing dissociation of SIRT1 from its endogenous inhibitor DBC1. Our experiments provide new insight on the in vivo mechanism of SIRT1 regulation and a new avenue for the development of pharmacological SIRT1 activators targeted at the dissociation of the SIRT1-DBC1 complex.”
The 2008 publication DBC1 is a negative regulator of SIRT1 reports: “The NAD-dependent protein deacetylase Sir2 (silent information regulator 2) regulates lifespan in several organisms1, 2, 3. SIRT1, the mammalian orthologue of yeast Sir2, participates in various cellular functions4, 5, 6, 7 and possibly tumorigenesis8. Whereas the cellular functions of SIRT1 have been extensively investigated, less is known about the regulation of SIRT1 activity. Here we show that Deleted in Breast Cancer-1 (DBC1), initially cloned from a region (8p21) homozygously deleted in breast cancers9, forms a stable complex with SIRT1. DBC1 directly interacts with SIRT1 and inhibits SIRT1 activity in vitro andin vivo. Downregulation of DBC1 expression potentiates SIRT1-dependent inhibition of apoptosis induced by genotoxic stress. Our results shed new light on the regulation of SIRT1 and have important implications in understanding the molecular mechanism of ageing and cancer.”
The DBC1–SIRT1 interaction increases following DNA damage and oxidative stress
The 2012 publication Regulation of SIRT1 activity by genotoxic stress reports: “SIRT1 regulates a variety of cellular functions, including cellular stress responses and energy metabolism. SIRT1 activity is negatively regulated by DBC1 (Deleted in Breast Cancer 1) through direct binding. However, how the DBC1–SIRT1 interaction is regulated remains unclear. We found that the DBC1–SIRT1 interaction increases following DNA damage and oxidative stress. The stress-induced DBC1–SIRT1 interaction requires the ATM-dependent phosphorylation of DBC1 at Thr 454, which creates a second binding site for SIRT1. Finally, we showed that the stress-induced DBC1–SIRT1 interaction is important for cell fate determination following genotoxic stress. These results revealed a novel mechanism of SIRT1 regulation during genotoxic stress.” So, there is another unvirtuous loop: if there is insufficient NAD+, the PARP DNA repair machinery will not work well creating greater oxidative stress which down-regulates SIRT1 further impairing the NAD salvage cycle.
Interestingly, it apears that resveratrol can block the SIRT1 down-regulating effect of DBC1/CCAR2.
The 2014 publication Resveratrol delays Wallerian degeneration in a NAD(+) and DBC1 dependent manner reports: “Axonal degeneration is a central process in the pathogenesis of several neurodegenerative diseases. Understanding the molecular mechanisms that are involved in axonal degeneration is crucial to developing new therapies against diseases involving neuronal damage. Resveratrol is a putative SIRT1 activator that has been shown to delay neurodegenerative diseases, including Amyotrophic Lateral Sclerosis, Alzheimer, and Huntington’s disease. However, the effect of resveratrol on axonal degeneration is still controversial. Using an in vitro model of Wallerian degeneration based on cultures of explants of the dorsal root ganglia (DRG), we showed that resveratrol produces a delay in axonal degeneration. Furthermore, the effect of resveratrol on Wallerian degeneration was lost when SIRT1 was pharmacologically inhibited. Interestingly, we found that knocking out Deleted in Breast Cancer-1 (DBC1), an endogenous SIRT1 inhibitor, restores the neuroprotective effect of resveratrol. However, resveratrol did not have an additive protective effect in DBC1 knockout-derived DRGs, suggesting that resveratrol and DBC1 are working through the same signaling pathway. We found biochemical evidence suggesting that resveratrol protects against Wallerian degeneration by promoting the dissociation of SIRT1 and DBC1 in cultured ganglia. Finally, we demonstrated that resveratrol can delay degeneration of crushed nerves in vivo. We propose that resveratrol protects against Wallerian degeneration by activating SIRT1 through dissociation from its inhibitor DBC1.”
Other relevant publications on DBC1/CCAR2 include:
- Deleted in breast cancer 1 (DBC1) protein regulates hepatic gluconeogenesis. (2014)
- DBC1 (Deleted in Breast Cancer 1) modulates the stability and function of the nuclear receptor Rev-erbα. (2013)
- Role of deleted in breast cancer 1 (DBC1) protein in SIRT1 deacetylase activation induced by protein kinase A and AMP-activated protein kinase. (2012)
- Deleted in breast cancer-1 regulates SIRT1 activity and contributes to high-fat diet-induced liver steatosis in mice. (2010)
Age-related decline in NAD+
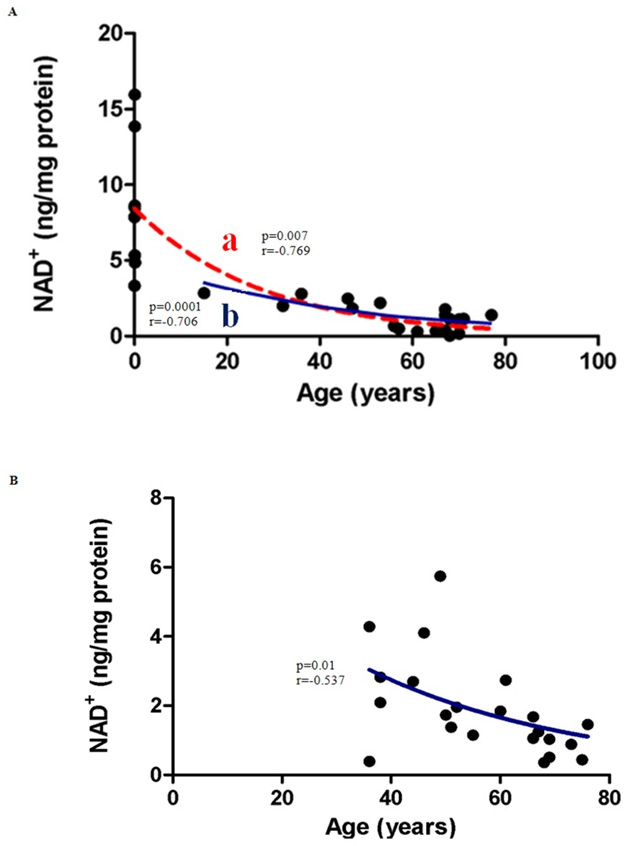
Image and legend source Correlation between NAD+ levels and Age in (A) Males (B) Females.. From: Age-Associated Changes In Oxidative Stress and NAD+ Metabolism In Human Tissue .
(A) NAD+ concentrations decline with age in males. NAD+ levels decreased significantly in males aged between 0–77 years (line a; p = 0.0007; n = 27). Pearson’s correlation coefficient for a normally distributed population, r = −0.769. The post-pubescent data for male subjects also showed a decline in NAD+ levels with age (line b; r = −0.706; p = 0.0001; n = 19). An exponential (first-order) least squares fit was used to generate the nonlinear trend lines (line a and b). (B)NAD+ concentrations decreased significantly with age (36–76) in post-pubescent females (p = 0.01; n = 22).Pearson’s correlation coefficient for a normally distributed population,r = −0.537. An exponential (first-order) least squares fit was used to generate the nonlinear trend line.”
NAD SYNTHESIS
in general, there are two major pathways via which NAD can be synthesized in the body: the de-novo pathway for synthesizing NAD from raw materials, and the salvage pathway which involves recycling NAD between its two states NAD+ and NADH.
The de-novo pathway.
Again from Dissecting Systemic Control of Metabolism and Aging in the NAD World: The Importance of SIRT1 and NAMPT-mediated NAD Biosynthesis: “NAD is synthesized from three major precursors—tryptophan, nicotinic acid, and nicotinamide [38] and [39]. Lower eukaryotes and invertebrates, such as yeast, worms, and flies, use nicotinic acid, a form of vitamin B3, as a major NAD precursor, whereas mammals predominantly use nicotinamide, another form of vitamin B3, for NAD biosynthesis. In mammals, NAMPT initiates the major NAD biosynthesis pathway by converting nicotinamide and 5′-phosphoribosyl-1-pyrophosphate (5′-PRPP) to nicotinamide mononucleotide (NMN), which is the rate-limiting step in this NAD biosynthesis [11] and [12]. The second enzyme, nicotinamide/nicotinic acid mononucleotide adenylyltransferase (NMNAT), completes NAD biosynthesis by transferring adenine from ATP to NMN. There are three distinct isoforms for NMNAT, NMNAT1-3, which are localized in nucleus, cytoplasm, and mitochondria, respectively, suggesting that NAD biosynthesis mediated by NAMPT and NMNAT might be compartmentalized in each subcellular compartment [40] and [41].” Later. we discuss how one possible strategy for enhancing body level of NAD is supplementation with NMN. This has worked remarkably well to produce health benefits in small animals, but the substance is now extremely expensive.
PROPERTIES OF THE NAD SALVAGE CYCLE
A diagram for the salvage pathway has already been offered above. the following diagram shows both the de-novo and the salvage pathway.
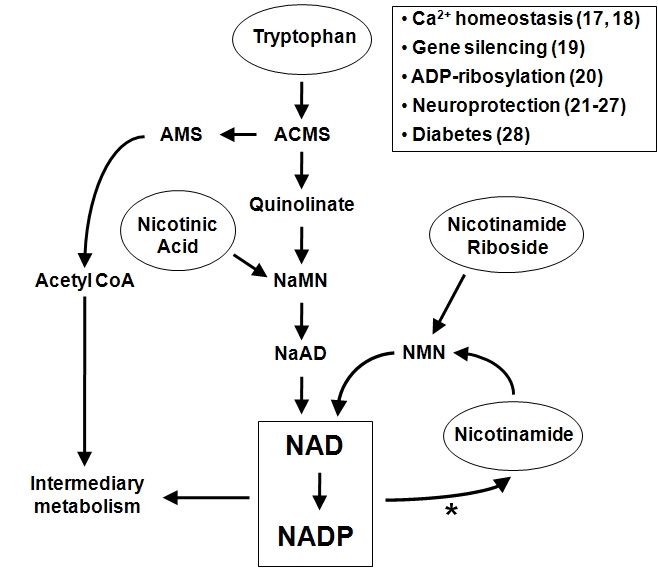
Important aspects of the salvage cycle are 1. The cycle is under circadian clock and SIRT1 control, 2. salvage cycle activities regulate expression of several genes via SIRT1 promoter activities, 3. ROS stress activating p53 is a regulator of the cycle, and 4. The NAD World includes extra-cellular actions as well as ones in different cell compartments. We expand on each of these points.
1. Circadian clock regulation of the NAD salvage cycle
The 2009 publication Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1 reports: “Many metabolic and physiological processes display circadian oscillations. We have shown that the core circadian regulator, CLOCK, is a histone acetyltransferase whose activity is counterbalanced by the nicotinamide adenine dinucleotide (NAD+)-dependent histone deacetylase SIRT1. Here we show that intracellular NAD+ levels cycle with a 24-hour rhythm, an oscillation driven by the circadian clock. CLOCK:BMAL1 regulates the circadian expression of NAMPT (nicotinamide phosphoribosyltransferase), an enzyme that provides a rate-limiting step in the NAD+ salvage pathway. SIRT1 is recruited to the Nampt promoter and contributes to the circadian synthesis of its own coenzyme. Using the specific inhibitor FK866, we demonstrated that NAMPT is required to modulate circadian gene expression. Our findings in mouse embryo fibroblasts reveal an interlocked transcriptional-enzymatic feedback loop that governs the molecular interplay between cellular metabolism and circadian rhythms.”
The 2010 publication Clocks in the NAD World: NAD as a metabolic oscillator for the regulation of metabolism and aging relates: ” Most recently, it has been demonstrated that SIRT1 regulates the amplitude and the duration of circadian gene expression through the interaction and the deacetylation of key circadian clock regulators, such as BMAL1 and PER2. More strikingly, we and others have discovered a novel circadian clock feedback loop in which both the rate-limiting enzyme in mammalian NAD biosynthesis, nicotinamide phosphoribosyltransferase (NAMPT), and NAD levels display circadian oscillations and modulate CLOCK:BMAL1-mediated circadian transcriptional regulation through SIRT1, demonstrating a new function of NAD as a “metabolic oscillator.” These findings reveal a novel system dynamics of a recently proposed systemic regulatory network regulated by NAMPT-mediated NAD biosynthesis and SIRT1, namely, the NAD World. In the light of this concept, a new connection between physiological rhythmicity, metabolism, and aging will be discussed.”
Here is a diagram of the situation:
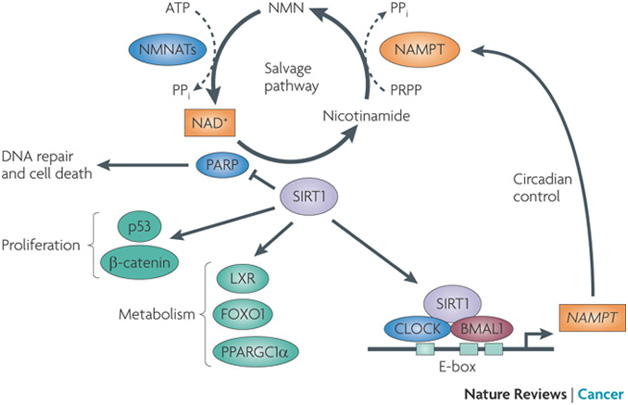
Image source ” The circadian clock controls the expression of nicotinamide phosphoribosyltransferase (NAMPT), which encodes the rate-limiting enzyme in mammalian NAD+ biosynthesis from nicotinamide. NAMPT catalyses the transfer of a phosphoribosyl residue from 5-phosphoribosyl-1-pyrophosphate (PRPP) to nicotinamide to produce nicotinamide mononucleotide (NMN), which is then converted to NAD+ by nicotinamide mononucleotide adenylyltransferases (NMNATs; there are three NMNAT genes). Oscillations in NAMPT levels result in circadian variations in NAD+ levels, which determines the activity of sirtuin 1 (SIRT1) and poly(ADP-ribose) polymerases (PARPs). Therefore, SIRT1 determines the oscillatory levels of its own coenzyme, NAD+ (Ref. 23). SIRT1 can also deacetylate and regulate proteins involved in metabolism and cell proliferation. Orange indicates circadian oscillation. FOXO1, forkhead box O1; LXR, liver X receptor; PPARGC1α, PPARγ co-activator 1α; PPi, pyrophosphate.”
Note that SIRT1 is intrinsic to the operation of the cycle, both regulating the cycle and being regulated by it.
Here is another Depiction of clock regulation of the salvage cycle via BMAL1 and CLOCK:
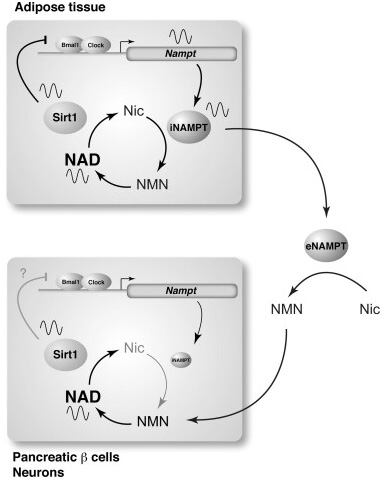
Image and legend source source ” A circadian oscillatory feedback loop regulated by NAMPT, SIRT1, and CLOCK/BMAL1 and a possible functional interplay between adipose tissue and two frailty tissues in the NAD World, pancreatic β cells and neurons. NAMPT and NAD levels display circadian oscillations that are regulated by CLOCK/BMAL1 in peripheral tissues, such as the liver and WAT. This NAD oscillation periodically activates SIRT1, which represses CLOCK/BMAL1-mediated transcription of clock target genes, including Nampt itself, completing an interlocked transcriptional-enzymatic feedback loop involving NAMPT-NAD and SIRT1-CLOCK/BMAL1. In pancreatic β cells and central neurons, intracellular NAMPT (iNAMPT) levels are so low [45] that they may not be able to drive the NAMPT–NAD–SIRT1-dependent circadian feedback loop. However, their NAD oscillation might be generated by incorporating NMN that is likely synthesized from nicotinamide (Nic) by extracellular NAMPT (eNAMPT) that could be periodically secreted from adipose tissue.”
2. The salvage pathway and SIRT1 regulation of gene expression.
NAD+ regulates gene expression via histone deacetylase activity of SIRT1 at gene promoter sites.
The 2009 publication Enzymes in the NAD+ salvage pathway regulate SIRT1 activity at target gene promoters reports: “In mammals, nicotinamide phosphoribosyltransferase (NAMPT) and nicotinamide mononucleotide adenylyltransferase 1 (NMNAT-1) constitute a nuclear NAD(+) salvage pathway which regulates the functions of NAD(+)-dependent enzymes such as the protein deacetylase SIRT1. One of the major functions of SIRT1 is to regulate target gene transcription through modification of chromatin-associated proteins. However, little is known about the molecular mechanisms by which NAD(+) biosynthetic enzymes regulate SIRT1 activity to control gene transcription in the nucleus. In this study we show that stable short hairpin RNA-mediated knockdown of NAMPT or NMNAT-1 in MCF-7 breast cancer cells reduces total cellular NAD(+) levels and alters global patterns of gene expression. Furthermore, we show that SIRT1 plays a key role in mediating the gene regulatory effects of NAMPT and NMNAT-1. Specifically, we found that SIRT1 binds to the promoters of genes commonly regulated by NAMPT, NMNAT-1, and SIRT1 and that SIRT1 histone deacetylase activity is regulated by NAMPT and NMNAT-1 at these promoters. Most significantly, NMNAT-1 interacts with, and is recruited to target gene promoters by SIRT1. Collectively, our results reveal a mechanism for the direct control of SIRT1 deacetylase activity at a set of target gene promoters by NMNAT-1. This mechanism, in collaboration with NAMPT-dependent regulation of nuclear NAD(+) production, establishes an important pathway for transcription regulation by NAD(+).”
3. NAD+ synthesis in the salvage cycle is in part mediated by ROS stress and p53 activation.
The 2014 publication The NAD+ synthesizing enzyme nicotinamide mononucleotide adenylyltransferase 2 (NMNAT-2) is a p53 downstream target reports: “NAD(+) metabolism plays key roles not only in energy production but also in diverse cellular physiology. Aberrant NAD(+) metabolism is considered a hallmark of cancer. Recently, the tumor suppressor p53, a major player in cancer signaling pathways, has been implicated as an important regulator of cellular metabolism. This notion led us to examine whether p53 can regulate NAD(+) biosynthesis in the cell. Our search resulted in the identification of nicotinamide mononucleotide adenylyltransferase 2 (NMNAT-2), a NAD(+) synthetase, as a novel downstream target gene of p53. We show that NMNAT-2 expression is induced upon DNA damage in a p53-dependent manner. Two putative p53 binding sites were identified within the human NMNAT-2 gene, and both were found to be functional in a p53-dependent manner. Furthermore, knockdown of NMNAT-2 significantly reduces cellular NAD(+) levels and protects cells from p53-dependent cell death upon DNA damage, suggesting an important functional role of NMNAT-2 in p53-mediated signaling. Our demonstration that p53 modulates cellular NAD(+) synthesis is congruent with p53’s emerging role as a key regulator of metabolism and related cell fate.
4. Action in the NAD World takes place in multiple cell compartments: the cytoplasm, the mitochondria and in the nucleus – and also in the plasma.
This point is illustrated in this diagram:
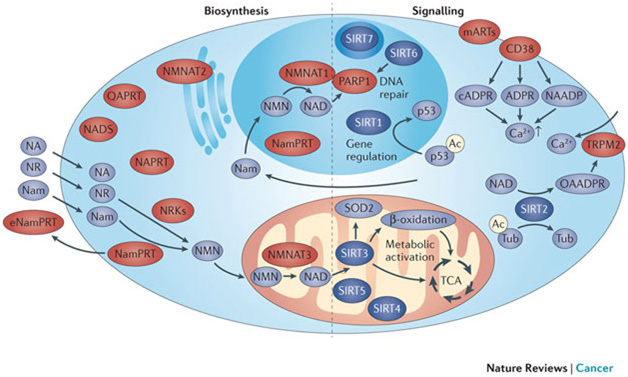
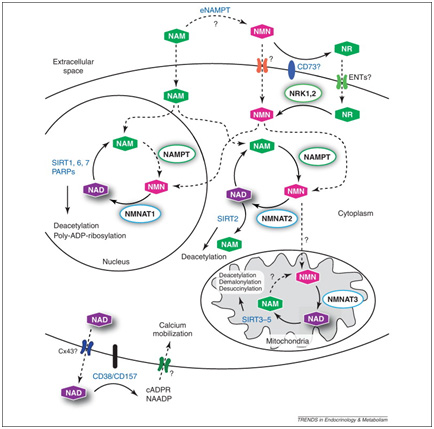
Image and legend source “The subcellular distribution of NAD+ biosynthetic enzymes in mammalian cells is shown on the left-hand side of the figure. All known biosynthetic reactions can take place in the cytosol, although conversion of nicotinamide (Nam) to NAD+ also takes place in the nucleus. However, NAD+ and its biosynthetic intermediates are likely to be freely exchangeable between the cytosol and the nucleus. Mitochondria contain nicotinamide mononucleotide (NMN) adenylyltransferase (NMNAT) activity and can therefore generate NAD+ from NMN. An extracellular form of Nam phosphoribosyltransferase (NamPRT), known as eNamPRT (also known as visfatin), is secreted from adipose tissue. Pyridine bases (Nam and nicotinic acid (NA)) and nucleosides (such as Nam riboside (NR)) enter cells by different transport mechanisms. The compartments of predominant NAD+ generation, the nucleus and mitochondria, also harbour the majority of intracellular NAD+-dependent signalling processes (right-hand side of the figure). Nuclear poly(ADP ribose) polymerase 1 (PARP1) generates polyADP ribose and probably accounts for the majority of nuclear NAD+ degradation. Direct interaction with NMNAT1 facilitates NAD+ supply to activated PARP1. Members of the sirtuin family are found in the nucleus, the cytosol and the mitochondria. SIRT1 is a key regulator of gene expression by deacetylating histone and non-histone targets such as p53. SIRT6 is also a nuclear protein. Among others, it activates PARP1, thereby facilitating DNA repair. SIRT7 is mostly localized to the nucleolus. SIRT2 localizes to the cytosol. SIRT3, SIRT4 and SIRT5 are located in mitochondria. SIRT3 is the major deacetylase in these organelles and a key regulator of metabolic pathways, including β-oxidation of fatty acids and the tricarboxylic acid cycle (TCA). Moreover, it deacetylates and activates superoxide dismutase 2 (SOD2), thereby suppressing reactive oxygen species (ROS) production. CD38, the main mammalian NAD+ glycohydrolase, and the majority of monoADP ribosyltransferases (mARTs) are found on the cell surface. All signalling reactions generate Nam as a common product, which can be recycled into the NAD+biosynthesis pathway by NamPRT.”
THE AVAILABILITY OF NAD+ IS CRITICAL FOR MITOCHONDRIAL FUNCTIONALITY
This has been known for a decade now. The 2004 publication Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition relates “—. In astrocytes, extracellular NAD(+) can raise intracellular NAD(+) concentrations. To determine whether NAD(+) depletion is necessary for PARP-1-induced MPT, NAD(+) was restored to near-normal levels after PARP-1 activation. Restoration of NAD(+) enabled the recovery of mitochondrial membrane potential and blocked both MPT and cell death. Furthermore, both cyclosporin A and NAD(+) blocked translocation of the apoptosis-inducing factor from mitochondria to nuclei, a step previously shown necessary for PARP-1-induced cell death. These results suggest that NAD(+) depletion and MPT are necessary intermediary steps linking PARP-1 activation to AIF translocation and cell death.”
The 2011 publication Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis reported: “During the last several years, evidence that various enzymes hydrolyze NAD into bioactive products prompted scientists to revisit or design strategies able to increase intracellular availability of the dinucleotide. However, plasma membrane permeability to NAD and the mitochondrial origin of the dinucleotide still wait to be clearly defined. Here, we report that intracellular NAD contents increased upon exposure of cell lines or primary cultures to exogenous NAD (eNAD). NAD precursors could not reproduce the effects of eNAD, and they were not found in the incubating medium containing eNAD, thereby suggesting direct cellular eNAD uptake. We found that in mitochondria of cells exposed to eNAD, NAD and NADH as well as oxygen consumption and ATP production were increased. Conversely, DNA repair, a well known NAD-dependent process, was unaltered upon eNAD exposure. We also report that eNAD conferred significant cytoprotection from apoptosis triggered by staurosporine, C2-ceramide, or N-methyl-N’-nitro-N-nitrosoguanidine. In particular, eNAD reduced staurosporine-induced loss of mitochondrial membrane potential and ensuing caspase activation. Of importance, pharmacological inhibition or silencing of the NAD-dependent enzyme SIRT1 abrogated the ability of eNAD to provide protection from staurosporine, having no effect on eNAD-dependent protection from C2-ceramide or N-methyl-N’-nitro-N-nitrosoguanidine. Taken together, our findings, on the one hand, strengthen the hypothesis that eNAD crosses the plasma membrane intact and, on the other hand, provide evidence that increased NAD contents significantly affects mitochondrial bioenergetics and sensitivity to apoptosis.”
Declining NAD+ can lead to the collapse of mitochondrially-mediated energy metabolism, The process appears to be reversible.
The December 2013 publication that catalyzed the current interest in NAD+ and mentioned at the begenning of this blog entry is Declining NAD+ Induces a Pseudohypoxic State Disrupting Nuclear-Mitochondrial Communication during Aging.
“Highlights
• A specific decline in mitochondrially encoded genes occurs during aging in muscle
• Nuclear NAD+ levels regulate mitochondrial homeostasis independently of PGC-1α/β
• Declining NAD+ during aging causes pseudohypoxia, which disrupts OXPHOS function
• Raising nuclear NAD+ in old mice reverses pseudohypoxia and metabolic dysfunction
“Ever since eukaryotes subsumed the bacterial ancestor of mitochondria, the nuclear and mitochondrial genomes have had to closely coordinate their activities, as each encode different subunits of the oxidative phosphorylation (OXPHOS) system. Mitochondrial dysfunction is a hallmark of aging, but its causes are debated. We show that, during aging, there is a specific loss of mitochondrial, but not nuclear, encoded OXPHOS subunits. We trace the cause to an alternate PGC-1α/β-independent pathway of nuclear-mitochondrial communication that is induced by a decline in nuclear NAD+ and the accumulation of HIF-1α under normoxic conditions, with parallels to Warburg reprogramming. Deleting SIRT1 accelerates this process, whereas raising NAD+ levels in old mice restores mitochondrial function to that of a young mouse in a SIRT1-dependent manner. Thus, a pseudohypoxic state that disrupts PGC-1α/β-independent nuclear-mitochondrial communication contributes to the decline in mitochondrial function with age, a process that is apparently reversible.”
” In oncology, the Warburg effect is the observation that most cancer cells predominantly produce energy by a high rate of glycolysis followed by lactic acid fermentation in the cytosol, rather than by a comparatively low rate of glycolysis followed by oxidation of pyruvate in mitochondria as in most normal cells.[4][5][6] The latter process is aerobic (uses oxygen). Malignant, rapidly growing tumor cells typically have glycolytic rates up to 200 times higher than those of their normal tissues of origin; this occurs even if oxygen is plentiful(ref).”
The following diagrams illustrate aspects of the NAD+ – mitochondrial connection:
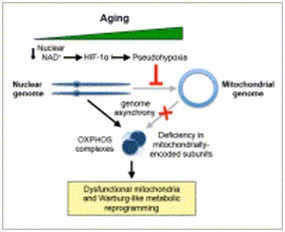
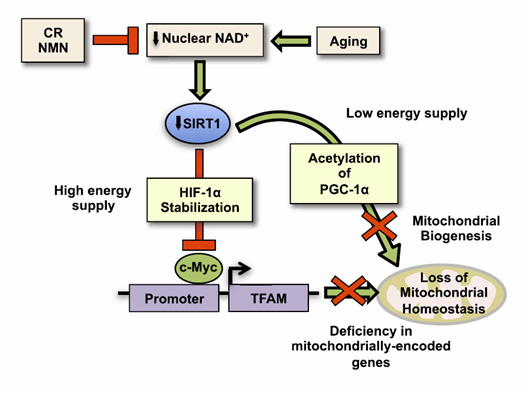
Image and legend source ” We show that, during aging, there is a specific loss of mitochondrial, but not nuclear, encoded oxphos subunits. We trace the cause to an alternate pgc-1a/b-independent pathway of nuclear-mitochondrial communication that is induced by a decline in nuclear NAD+ and the accumulation of hif-1a under normoxic conditions, with parallels to warburg reprogramming. Deleting sirt1 accelerates this process, whereas raising NAD+ levels in old mice restores mitochondrial function to that of a young mouse in a sirt1-dependent manner. Thus, a pseudohypoxic state that disrupts PGC-1a/b-independent nuclear-mitochondrial communication contributes to the decline in mitochondrial function with age, a process that is apparently reversible.”
The following two diagrams illustrates matters discussed above that go on in key cell compartments and outside the cell:
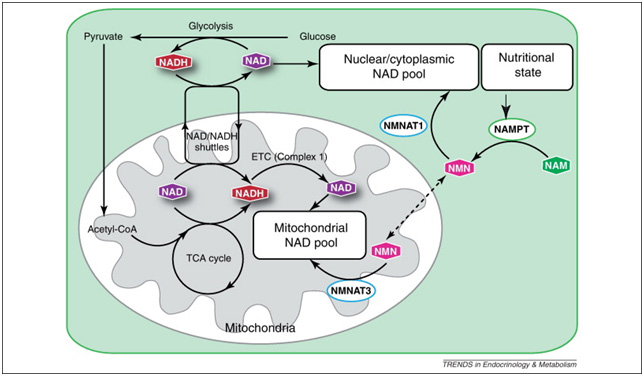
Images and legend source ” Maintenance of the mitochondrial NAD pool. Although separate, the mitochondrial and nuclear/cytoplasmic NAD pools are intricately connected through the NAD/NADH-redox shuttles (most commonly the malate/aspartate and the glycerol-3-phosphate shuttles) and NAD biosynthetic pathways in each subcellular compartment. Multiple cellular processes play an important role in maintaining an optimal NAD/NADH ratio between mitochondria and the cytoplasm, including glycolysis, the tricarboxylic acid (TCA) cycle, and oxidative phosphorylation by the electron transport chain (ETC). Mitochondrial and nuclear/cytoplasmic NAD biosynthetic pathways are balanced in response to nutritional and environmental stimuli.”
Why this is all important for longevity is illustrated by these diagrams for mice:
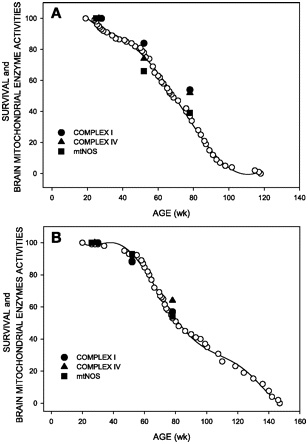
“Activity of brain mitochondrial enzymes that are markers of aging in relation to mice survival. Complex I, complex IV, and mitochondrial nitric oxide (NO) synthase (mtNOS) activities in male (A) and female (B) mice.”
More to come
This blog entry has covered some important basics related to the NAD world, but there is a great deal more to say about the topic. There will be at least two more blog entries in this series. Part 2 concentrates on the reasons for focusing on NAD+, particularly with respect to interventions that are seriously likely to lead to longer healthier lives. The Part 3 blog entry describes 30 Major Factors that Control SIRT1 expression, SIRT1 Activity, and SIRT1-mediated Aging. An anticipated Part 4 blog entry will discuss various associated topics such as the relationship of SIRT1 and inflammation, causes of the Warburg Effect, and possible health interventions that are designed to improve the NAD+/NADH ratio.

{kind=link}
{kind=link}
Hi Vince.
I have put many hours into studying NAD and it’s metabolites, particularly the use of Nicotinic acid to increase NAD levels. So far as I am aware the enzymatic step for converting Nicotinic acid into Nicotinamide is not present in mammals. That is not necessarily the case in other organisms, but appears to be the case in mammals. I currently take 500 mg of Nicotinic acid per day to this end. That said I have many issues with Nicotinic acid, mainly those centering around toxicity and it’s efficiency at raising NAD levels. Nicotinic acid also seems to be tissue specific. Anyway, could you please let me know if I am mistaken about the conversion of Nicotinic acid into Niacinamide.
Many thanks
Phil
Thanks Phil
I will have another look and get back here again soon
Vince
Pingback: Restoring Mitochondrial NAD/ NADH Ratio - AWAREmed Addiction Training
Hi Vince
Thank you for responding to my post.
After reading your comment on Niacin and it’s conversion to Niacinamide I decided to pursue matters further. After some hours of digging I discovered that nicotinic acid is converted to nicotinamide via nicotinic acid mononucleotide to form the nicotinic acid analogue of NAD. NAD is then enzymatically cleaved to form nicotinamide. The intestinal mucosa and liver are apparently rich in niacin conversion enzymes, such as NAD glycohydrolase, which releases nicotinamide from NAD. Nicotinamide appears to be the predominant circulating form of the vitamin.
Given this insight I will stop taking Nicotinic acid as there appears to be no advantage to its use.
I think in my case, a little knowledge is dangerous. I spent far too much time studying the de novo and salvage pathways and did not notice that there are NAD consuming enzymes.
I look forward to your next post on the subject and will study its contents with interest. In the mean time I suppose it’s back to trying to reduce the rate of accruing DNA damage (Blackburn-Ornish, Bruce Ames etc). Nicotinamide Riboside is prohibitively expensive to me, particularly if I plan on taking it in pharmacologic dosages.
Kind regards
Phil
http://www.ncbi.nlm.nih.gov/pubmed/6218262
Not sure about humans by in rats supplementing nicotinic riboside is a waste of time
I mean money since its absorbed as nicotinamide
Dear Vince
http://www.jbc.org/content/285/53/41211.short
Axonal Degeneration Is Blocked by Nicotinamide Mononucleotide Adenylyltransferase (Nmnat) Protein Transduction into Transected Axons
NMN accumulation in the axons degenerates the axons. Is this contradictory to what we assume?
Pingback: NAD+ an emerging framework for life health and life extension — Part 2: Deeper into the NAD World, hopeful interventions | AGING SCIENCES – Anti-Aging Firewalls
Pingback: A simple, comprehensive plan to prevent or reverse Alzheimer’s Disease and other neurodegenerative diseases – Part 1: The Plan | AGING SCIENCES – Anti-Aging Firewalls
I just want to clarify about what you said about cdk-1 pathway and cell division. So your saying Sirt1 turns on cdk-1 which increases cell division and also stimulates growth hormone and igf1? I just want to make this clear because im taking NAD+ and i dont want to grow a bigger body or face than what i already have. Im 21 and im looking to stay my age or younger for a hundred years or so… Im thinking increasing cell division might work against this. My goal is to actually reverse cell division if possible. Thank you.
Pingback: Digital Health Part 4: Heart rate stress biomarkers derivable from smart watch data | AGINGSCIENCES™ – Anti-Aging Firewalls™
Hi, guys–
Like others, I’m a little confused by your statements about niacin being converted in the body to nicotinamide. The reference you give to the nicotinamide Wikipedia page suggests that niacin is directly converted to nicotinamide “in vivo”, but I don’t find a pathway for that.
By contrast, the Wikipedia page for niacin states “Niacin cannot be directly converted to nicotinamide, but both compounds are precursors of the coenzymes nicotinamide adenine dinucleotide (NAD) and nicotinamide adenine dinucleotide phosphate (NADP) in vivo.[4] ”
NAD itself may eventually yield nicotinamide, but it looks to me as if that is the case whether the original source is niacin, nicotinamide riboside, or nicotinamide mononucleotide.
The pathways I see are
1) Niacin (Na)—>NaMN—>NaAD—>NAD+
2) Nicotinamide (Nam)—>NMN—>NAD+
3) NR—>NMN—>NAD+
and then, of course, NAD+ can be broken down and recycled via Nam salvage. But it looks to me as if you end up with extra Nam whether you take Niacin or NR or Nam or even NMN.
Could you please clarify why you believe Niacin produces more Nam per molecule than NR does? I’m not saying it doesn’t. I’m just saying I don’t see the pathway.
Pingback: Part 5 of the NAD world: the conflicting roles of NAMPT – inflammation or rescue? Also Part 1 on a new series on inflammation | AGINGSCIENCES™ – Anti-Aging Firewalls™
Pingback: How to Increase Your Chances of Living Forever – The Epoch
Pingback: CHRONOBIOLOGY OF TISSUE REGENERATION - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™