
By James P Watson with commentary and assistance by Vince Giuliano
Introduction by Vince Giuliano
This blog entry relates another important story of what goes on in the NAD world. This is also the first entry of what I expect to be several having to do with inflammation. To quote Jim Watson, “The most important three things about aging are inflammation, inflammation, and inflammation. Inflammation may be due to multiple causes, exist in multiple forms, and is a common factor to the vast majority of diseases including those most closely associated with aging. Here is the story we will document here:
“There once was (and still is) a wonderful substance that players in the anti-aging game like us want to have more of in our cells because it makes us healthy and maybe even live longer. Called NAD+, publications were written about this great substance by the best of researchers in the greatest temples of longevity research (and, by us in this blog).
And supplement makers started selling expensive pills that could trick our cells into making more of this. One of the critical molecules in your cells responsible for making NAD+, a close cousin molecule, is called NAMPT, so people in the know started wanting to have more of that too.
But then, a dark side of all of this begin to emerge as it was realized that NAMPT has long been known to be a traveler using various aliases, especially outside of cells where it did shocking things. NAMPT was often behaving inappropriately for its princely role when it is outside our cells. Not to be trusted, extra-cellular NAMPT (eNAMPT) can be caught creating horrible inflammation and is closely associated with highly disreputable diseases. Oozing out of fat cells and helping create colorectal cancer is not politically correct for a supposedly good guy. The same is true for destroying cartilage in both osteo and rheumatoid arthritis. We have written before about the good side of NAMPT when it is in cells. This present blog entry is an expose of the bad inflammation-generating side of eNAMPT. And, it turns out that eNAMPT is not completely bad, after all.”
This blog entry is concerned with one particular cause of inflammation associated with eNAMPT (extracellular NAMPT, nicotinamide phosphoribosyltransferase). This molecule has a dual character and a history of aliases. It is also known as Visfatin and as Pre-B Cell Colony Enhancing Factor.
We recall NAMPT is a rate-limiting enzyme in the biosynthesis of NAD+ (nicotinamide adenine dinucleotide), an essential cellular coenzyme which we generally want to see promoted. We have previously focused on the intra-cellular role of NAMPT which is as an enzyme in the pathway responsible for the generation of NAD. NAMPT generates nicotinamide mononucleotide NMN from nicotinamide. NMN is then converted to NAD via nicotinamide riboside (NR). How this all works is all laid out in first four blog entries related to the NAD world:
The Part 1 blog entry in the NAD World series provided an overview treatment of the NAD World and its nuances: to identify the major molecular entities involved, their roles, health and longevity ramifications, the reasons for the current excitement, and to begin to clarify what is actually known and what the remaining uncertainties are.
The Part 2 blog entry in the NAD World series concentrates on the reasons for focusing on NAD+, particularly with respect to interventions that are seriously likely to lead to longer healthier lives. It discusses molecular processes in the NAD salvage cycle that are responsible for the health-inducing and life-extending properties of calorie restriction, and further discuss the key roles of Sirtuins, SIRT1, SIRT6 and SIRT7 in particular.
The Part 3 blog entry in the NAD World series identifies 30 Major Factors that Control SIRT1 Expression, SIRT1 Activity, and SIRT1-mediated Aging.I. The NAD+/NADH ratio and what affects it.
The Part 4 blog entry in the NAD World series is concerned with the NQ01 gene, the Warburg effect, SIRT 1 and inflammation, and possible interventions.
Most of the concern in these blog entries has been with the roles of iNAMPT (intracellular NAMPT) which functions as an enzyme.
The extra-cellular role of NAMPT is a whole different story of concern in this current blog entry where e-NAMPT functions not only as an enzyme but also as a pro-inflammatory cytokine.
This blog entry is also Part 1 of a new blog series on Inflammation, concerned specifically with eNAMPT-related inflammation. Part 2 of that series is the Tale of three stress-sensors and their-interactions: 1. inflammation, 2. genomic-instability (p53). and 3. oxidative stress (nrf2). Part 3 if this series is concerned with resolving inflammation, and the important roles of certain mediators, namely resolvins, protectins, maresins and lipoxins. These are found in fish oil! Part 4 of the inflammation series is concerned with an amazingly effective new approach for averting the biggest killers of older people, atherosclerosis and heart disease – that being inhibition of the PCSK9 protein. Other blog entries in this series will be forthcoming.
On to the Jim Watson entry
I have been uncovering more and more paradoxical factors associated with NAMPT. eNAMPTappears to be both a “bad thing” and a “good thing”. The difference may have to do with the dose (i.e. Biphasic dose-response curve) or it may have to do with the zip code (i.e. What cell or what tissue), or eNAMPT may have what is called “pleiotropic effects”. What is clear to me is this fundamental fact: eNAMPT has both a “cytokine effect” and an “enzyme effect”. The inhibitor FK866 and NMN inhibit the “enzyme effect” but do not inhibit the “cytokine effect”.
The “cytokine effect” is mostly pro-inflammatory, but also promotes survival of cells such as cancer cells and macrophages. The “enzymatic effect” may indirectly affect inflammation/immunity by providing NMN substrates for conversion to NR, and subsequent plasma membrane uptake by cells that are NAD deficient (NMN does not appear to be taken up directly, but is usually converted into NR which is absorbed).
What is upsetting the apple cart even more is the fact that much of the “dogma about biomarkers of aging and biomarkers of cellular senescence” may be wrong! Specifically, the Japanese as well as others showed that IL-6, TNF-alpha, and CRP all predict all cause mortality and lifespan more accurately than age or gender. However, I have found that all of these 3 “biomarkers of aging” are triggered by increased levels of eNAMPT (IL-6, TNF-a, and CRP). Likewise, the classic biomarkers of cellular senescence (I.e. The SASP components…IL-6, IL-8, MCP-1, etc.) are all expressed from NON-SENESCENT CELLS in response to eNAMPT signaling! This makes me question the use of these cytokines as biomarkers for aging and biomarkers for cellular senescence, since their levels are easily altered by eNAMPT expression! We know that eNAMPT expression is NOT the entire picture of aging (aging includes DNA damage/mutations, epigenetic dysregulation, cellular senescence, increased mTOR signaling, mitochondrial dysfunction, proteotoxicity, autophagy failure, etc).
This raises many questions about the validity of inflammatory cytokines as true biomarkers of aging, since eNAMPT activity/signaling can be inhibited by NMN and by FK866. It also calls into question the link between cellular senescence and these same cytokines (IL-6, IL-8, MCP-1). Here are some more facts about eNAMPT and a list of the proteins that are secreted by cells in response to eNAMPT signaling.
This quote from the 2010 publication Pre-B Cell Colony Enhancing Factor/NAMPT/Visfatin in Inflammation and Obesity- Related Disorders telegraphs some key points which will be covered here: “Whereas prototypic adipocytokines such as adiponectin or leptin are mainly derived from adipocytes, others such as pre-B cell colony enhancing factor (PBEF)/nicotinamide phosphoribosyl transferase (NAMPT)/visfatin or resistin are produced by various cell types throughout the body. Although first discovery of this molecule as PBEF suggested primarily a cytokine function, its rediscovery as the key enzyme in nicotinamide adenine dinucleotide (NAD) generation has considerably widened its biological perspective. Finally, the same molecule was introduced as visfatin claiming an insulin-mimetic effect which has been questioned. Both extracellular (cytokine-like) and intracellular (enzymatic) functions are responsible for its relevance in immune, metabolic and stress responses. Its cytokine functions are mainly pro-inflammatory as it induces potently various other pro-inflammatory cytokines such as tumor necrosis factor alpha (TNFα) or interleukin-6 (IL-6). Its intracellular functions concentrate on the regulation of the activity of NAD-consuming enzymes such as various sirtuins thereby also affecting TNFα biosynthesis, cell life-span and longevity. Biochemical neutralization of PBEF/NAMPT/visfatin has been proven effective in various models of inflammation including sepsis/arthritis and in various models of cancer. Patients with non-alcoholic fatty liver disease (NAFLD) exhibit increased serum concentrations of PBEF/Nampt/visfatin and weight loss is associated both with a decrease in serum levels and reduced liver expression. Many of the biological functions of this “cytokine-enzyme” have been characterized in the last years, however, its definite role in various metabolic, inflammatory and malignant diseases has yet to be defined.”
- The pro-inflammatory effects of eNAMPT
One of the most unusual molecules in nature is the secreted form of NAMPT, called “eNAMPT,” eNAMPT is secreted in response to inflammation and also causes inflammation. Thus it is both a “cause” and an “effect” of inflammation. Many other names have been attached to eNAMPT before they were all found to be the same molecule. They include “Pre-B cell colony enhancing factor (PBEF) and Visfatin. (Here we shall just call it eNAMPT). eNAMPT increases circulating levels of pro-inflammatory cytokines, including IL-6, TNF-alpha, IL-1B, and TGF-B1. It also increases the chemokine receptor CCR3, VEGF, VEGFR, and MCP-1. eNAMPT activates the Toll-like receptor 4 (TLR4) in a unique way, triggering NF-kB mediated gene expression in cells. In conclusion, eNAMPT may be the link between visceral fat and systemic inflammation. It also may be the major molecular mechanism behind “sterile inflammation” and “inflammaging”, which are ways of describing the phenomena seen with aging and age-related diseases.
References:
2013 Pre-B cell colony enhancing factor (PBEF), a cytokine with multiple physiological functions “Extracellular PBEF has been shown to increase inflammatory cytokines, such as TNF-α, IL-1β, IL-16, and TGF-β1, and the chemokine receptor CCR3. PBEF also increases the production of IL-6, TNF-α, and IL-1β in CD14+ monocyctes, macrophages, and dendritic cells, enhances the effectiveness of T cells, and is vital to the development of both B and T lymphocytes.”
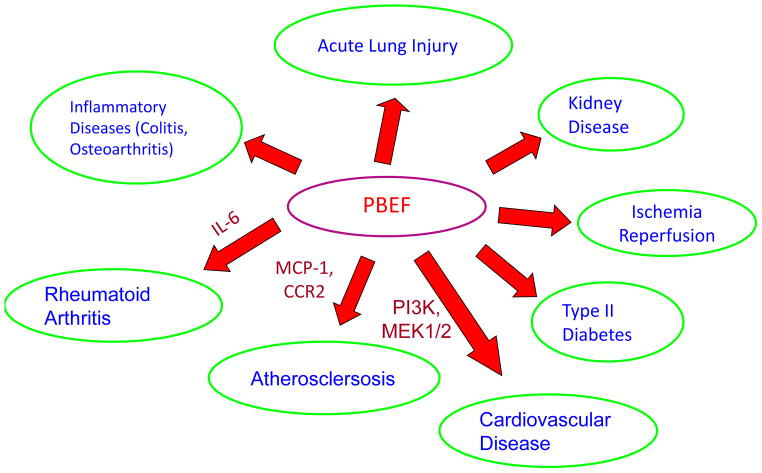
Image source Note that eNAMPT creates these terrible problems traveling here when using the alias PBEF
2009 Nicotinamide phosphoribosyltransferase (Nampt): A link between NAD biology, metabolism, and diseases “New interest in NAD biology has recently been revived, and enzymes involved in NAD biosynthetic pathways have been identified and characterized in mammals. Among them, nicotinamide phosphoribosyltransferase (Nampt) has drawn much attention in several different fields, including NAD biology, metabolism, and immunomodulatory response. The research history of this protein is peculiar and controversial, and its physiological function has been a matter of debate. Nampt has both intra- and extracellular forms in mammals. Intracellular Nampt (iNampt) is an essential enzyme in the NAD biosynthetic pathway starting from nicotinamide. On the other hand, an extracellular form of this protein has been reported to act as a cytokine named PBEF, an insulin-mimetic hormone named visfatin, or an extracellular NAD biosynthetic enzyme named eNampt. This review article summarizes the research history and reported functions of this unique protein and discusses the pathophysiological significance of Nampt as an NAD biosynthetic enzyme vs. a potential inflammatory cytokine in diverse biological contexts.”
- eNAMPT is secreted by neutrophils, microglia, macrophages, and visceral fat cells when these cells are stimulated by LPS or pro-inflammatory cytokines.
Pro-inflammatory cytokines trigger eNAMPT and eNAMPT triggers the gene transcription of more pro-inflammatory cytokines from the neurotrophils, macrophages, microglia, and visceral fat. With LPS stimulation of neutrophils, eNAMPT expression occurs 5 hours later. This is what is classically called a “positive feedback loop” in molecular biology, or could be called a “self-perpetuating fate” in theology. Regardless of the description, eNAMPT becomes a mediator of ongoing, chronic inflammation, long after the original trigger of inflammation is gone. This has often been referred to as “sterile inflammation”.
References:
From the book 2016 The Stress Response of Critical Illness: Metabolic and Hormonal Aspects
- eNAMPT has a protein sequence that “mimics” bacterial lipopolysaccharide (LPS) which triggers Toll-like receptor 4 signaling.
Lipopolysaccharide (LPS) is the molecular name for bacterial endotoxin. When lab experimenters want to create inflammation in a mouse or rat, they usually use LPS to do that. LPS triggers inflammation by binding to cells via the mammalian cell Toll-like receptor 4 (TLR4). Although early reports suggested that TLR2 also mediated LPS toxicity, more up-to-date findings have confirmed that TLR4 is the sole molecular receptor for LPS. TLR4 receptor triggering leads to the secretion of many pro-inflammatory, pro-angiogenic, and anti-apoptotic compounds from neutrophils, activated lymphocytes, macrophages, and visceral fat cells.
References:
2004 Tlr4: central component of the sole mammalian LPS sensor.
2012 Regulation of neutrophil function by NAMPT
- Inflammation up-regulates eNAMPT and eNAMPT up regulates inflammation.
In experimental studies where cells are activated by LPS, many genes are upregulated via NF-kB. One of these is NAMPT, which is transcribed, translated, and secreted from the cell as eNAMPT. Thus eNAMPT can be considered both the “cause” and the “effect” of Inflammation. Since bacterial infections (LPS) are common triggers of inflammation, eNAMPT can be consIdered an “amplifier of infectious inflammation” or in the absence of LPS, eNAMPT can be considered a mediator of “sterile inflammation.” Unfortunately, chronic inflammation acts as a “damper” on NAD production in the liver, in adipose tissue, in the pancreatic beta-islet cells, and in the brain. Thus the “true enemy” is inflammation, NOT eNAMPT! The diagram below from Imai’s recent 2013 paper illustrates this well.
References:
2012 Regulation of neutrophil function by NAMPT
2013 The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing
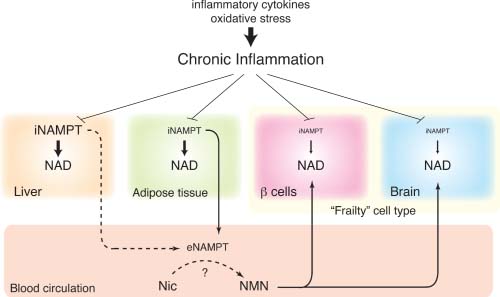
Image and legend source. “The concept of the NAD World and the possible effect of chronic inflammation. Pancreatic β-cells and neurons (the brain) are two major frailty points in the NAD World because these two cell types have very low levels of intracellular nicotinamide phosphoribosyltranferase (iNAMPT). These particular cell types likely depend on extracellular nicotinamide mononucleotide (NMN), which is speculated to be synthesized by extracellular nicotinamide phosphoribosyltranferase (eNAMPT) secreted by adipose tissue, and maintain optimal nicotinamide adenine dinucleotide (NAD) levels for their functions. Chronic inflammation, which is caused by inflammatory cytokines and oxidative stress, decreases NAMPT and NAD levels in multiple tissues, contributing to the pathogenesis of age-associated metabolic complications, such as type 2 diabetes. It still remains unclear whether chronic inflammation in adipose tissue also decreases plasma eNAMPT levels and remotely affects the functions of ‘frailty’ cell types.” So is eNAMPT good or bad? Like many other key molecules we have studied, the answer appears to be “both.”
- TLR4 signaling by eNAMPT triggers both NF-kB and AP-1 mediated signaling.
Whereas NF-kB is the classic “master switch” for inflammatory genes, AP-1 is a lesser known transcription factor that also triggers inflammatory genes. Because of the dual transcription factor signaling pathways for eNAMPT, blocking NF-kB alone with molecules like steroids or certain phytosubstances will fail to fully block the pro-inflammatory and pro-angiogenic effects of eNAMPT signaling. Of course, certain phytosubstances like curcumin do block both NF-kB and AP-1 inflammation(ref).
Reference: 2007 Pre-B-cell Colony-enhancing Factor (PBEF/Visfatin) Gene Expression is Modulated by NF-κB and AP-1 in Human Amniotic Epithelial Cells
- SIRT1 deacetylates iNAMPT, which enhances NAMPT activity and drives the secretion of eNAMPT from visceral fat cells.
This is one of the “paradoxical aspects” of the eNAMPT story. SIRT1 is typically thought of as a CR pathway enzyme that has anti-inflammatory effects. But here SIRT1 is playing a positive role in the secretion of eNAMPT from fat cells by deacetylation of lysine 53 (K53) on the NAMPT protein. This is the “secrete me” signal that triggers the export of the deacetylated eNAMPT out of the cell. In the plasma, fat cell-derived eNAMPT secretion circulates systemically and affects the hypothalmus. In the hypothalmus, the circulating eNAMPT increases NAD+ in the hypothalmus. Decreased eNAMPT decreases NAD+ in the hypothalmus, whereas increased eNAMPT in the plasma increases NAD in the hypothalmus. In NAMPT knock-out mice, NMN “rescues” the defect and the mice become more physically active. This is very puzzling until you put it in the context of fasting (CR) vs times of food abundance. Specifically, iNAMPT is normally acetylated at lysine 53 in both white fat and brown fat. However with fasting or starvation, SIRT1 deacetylates iNAMPT at lysine 53 (K53) which promotes its export. Thus fasting is an iNAMPT export signal leading to eNAMPT and its possible sequela as outlined here. Another example of the multiple good-bad roles played by eNAMPT.
References:
2015 SIRT1-Mediated eNAMPT Secretion from Adipose Tissue Regulates Hypothalamic NAD+ and Function in Mice
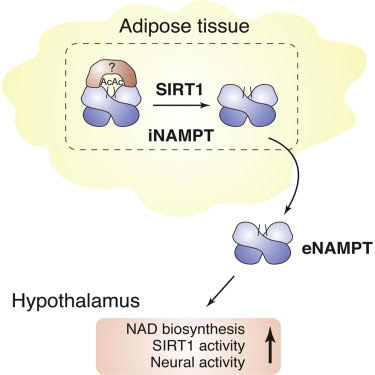
Image and legend source. “Nicotinamide phosphoribosyltransferase (NAMPT), the key NAD+ biosynthetic enzyme, has two different forms, intra- and extracellular (iNAMPT and eNAMPT), in mammals. However, the significance of eNAMPT secretion remains unclear. Here we demonstrate that deacetylation of iNAMPT by the mammalian NAD+-dependent deacetylase SIRT1 predisposes the protein to secretion in adipocytes. NAMPT mutants reveal that SIRT1 deacetylates lysine 53 (K53) and enhances eNAMPT activity and secretion. —“
- eNAMPT triggers systemic Insulin resistance and IGF-1 receptormediated PGE2-induced arthritis
The link between insulin resistance and eNAMPT is among the most fascinating things I have learned recently. This finally explains the links between fat (eNAMPT source), the regulation of the Insulin/IGF-1 pathway, and NMN. For quite some time, eNAMPT (also called PBEF or Visfatin) has been linked to inflammation, but exactly how eNAMPT was linked was unclear. This has largely been cleared up in the last 3-4 years.
- eNAMPT, IGF-1 signaling, and inflammation
In 2012, Jacques and colleagues from Marie Curie University in Paris showed that in joints, eNAMPT triggers inflammation by inducing PGE2 synthesis by chondrocytes. In cells with no IGF-1 receptors, eNAMPT triggered PGE2 biosynthesis. In cells with two functional copies of the IGF-1R, eNAMPT did not trigger nearly as much PGE2 release. However, Jacques and colleagues were not able to show if eNAMPT regulated the insulin receptor. They did show that inhibiting eNAMPT with FK866 gradually decreased PGE2 release and administering exogenous Nicotinamide increased PGE2 release.
Reference: 2012 Proinflammatory actions of visfatin/nicotinamide phosphoribosyltransferase (Nampt) involve regulation of insulin signaling pathway and Nampt enzymatic activity “We conclude that the proinflammatory actions of visfatin in chondrocytes involve regulation of IR signaling pathways, possibly through the control of Nampt enzymatic activity.”
- eNAMPT is the molecular cause of Insulin Resistance
The exact mechanism of how fat-induced inflammation causes insulin resistance has long remained a mystery. The mystery may have been solved in 2015 when a large, multi-center research group from Canada and China showed that eNAMPT induces the translocation if the insulin receptor out of lipid micro domains (lipid rafts) into non-lipid raft regions of the cell membrane. This had the net effect of making the insulin receptor resistant to insulin signaling (I.e. It did not trigger Akt phosphorylation). This effectively reduced insulin signaling by moving the IR to the non-raft region, which caused cells to become insulin resistant. This has explained the mystery of why insulin receptor density in cells does not decrease with fat-Induced inflammation, but that insulin resistance still occurs with fat-induced inflammation.
References:
2015 Pre-B cell colony enhancing factor induces Nampt-dependent translocation of the insulin receptor out of lipid microdomains in A549 lung epithelial cells “We conclude that PBEF can inhibit insulin signaling through the IR by Nampt-dependent promotion of IR translocation into the nonraft domains of A549 epithelial cells. PBEF-induced alterations in the spatial geometry of the IR provide a mechanistic explanation for insulin resistance in inflammatory states associated with upregulation of PBEF.”
2013 Pre-B cell colony enhancing factor (PBEF), a cytokine with multiple physiological functions
Conclusion re, eNAMPT and insulin resistance
The circulating molecule by which obesity (mainly visceral fat) can induce insulin resistance in distant organs (muscle) has now been identified – it is eNAMPT. eNAMPT is secreted mainly by visceral fat and inflammatory cells and circulates in the bloodstream as an enzyme and a pro-inflammatory, pro-angiogenic cytokine. In distant cells (such as muscle), it triggers insulin receptor movement from lipid rafts to non-lipid raft regions, effectively inactivating the intracellular signaling cascade that centers around Akt phosphorylation. Thus eNAMPT is the molecular “cause” of insulin resistance. eNAMPT also plays a role in triggering bioactive lipid-mediated inflammation (I.e. PGE2) in inflammatory diseases such as arthritis. For eNAMPT to trigger PGE2 release, there must be IGF-1 receptors (IGF1R) present. IGF1Rs are required for full eNAMPT-mediated PGE2 release. This IGF-1 and IGF1R play vital roles in the mechanism by which eNAMPT triggers PGE2 release in arthritis. This effect can be blocked with FK866, an eNAMPT inhibitor, or NMN, which inhibits eNAMPT by “feedback inhibition”.
- IL-8
IL-8 is secreted by the eNAMPT target cell by eNAMPT binding to the TLR4 receptor on the target cell and triggering IL-8 gene expression.
IL-8 is a classic biomarker of cellular senescence, proposed by Mayo Clinic and Buck Institute researchers. However, mRNA for IL-8 is increased 2-8 fold with eNAMPT over-expression in cells that are NOT senescent. The cell surface receptor activated by eNAMPT on the target cell is the Toll-like receptor 4 (TLR4). The two transcription factors that mediate this eNAMPT effect in the target cell are NF-kB and AP-1, classical villains of unwanted inflammation. These are not the transcription factors that trigger IL-8 secretion in senescent cells. (There it is JAK/STAT signaling pathway). Thus IL-8 may NOT always be due to senescent cell secretion….it may just be due to eNAMPT binding to a non-senescent cell and triggering AP-1 mediated IL-1B secretion, which then triggers IL-8.
References:
2008 A critical role of PBEF expression in pulmonary cell inflammation and permeability
2002 Pre-B-cell colony-enhancing factor, a novel cytokine of human fetal membranes
- IL-6
eNAMPT triggers the production of IL-6 in target cells in humans.
References:
2002 Pre-B-cell colony-enhancing factor, a novel cytokine of human fetal membranes
2010 Pre-B Cell Colony Enhancing Factor/NAMPT/Visfatin in Inflammation and Obesity- Related Disorders
- IL-1Beta
IL-Beta expression is triggered by eNAMPT
eNAMPT also triggers IL-1Beta secretion by cells. (IL-1B actually then triggers IL-8 secretion). While IL-1B is not a classic biomarker for cellular senescence, it is probably the #1 cytokine associated with osteoarthritis (OA), rheumatoid arthritis (RA), and many other chronic inflammatory diseases. Is OA and RA inflammation due to eNAMPT? Probably not! However I have read some articles that suggest that eNAMPT does play a role in arthritis. Here ls one:
Reference: 2004 Pre–B cell colony–enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis
- IL-16
IL-16 expression triggered by eNAMPT
eNAMPT signaling also stimulates IL-16 gene expression and production of the IL-16 cytokine. Here there appears to be a clear difference from cellular senescence. With cellular senescence, there is an increase in IL-6 and other cytokines, but the literature related to senescence-stimulated cytokines does not appear to mention IL-16 to our knowledge.
References:
2008 A critical role of PBEF expression in pulmonary cell inflammation and permeability
- CCR3
CCR3 expression triggered by eNAMPT
CCR3 is a chemokine
References:
2008 A critical role of PBEF expression in pulmonary cell inflammation and permeability “PBEF expression also affected the expression of two other inflammatory cytokines (IL-16 and CCR3 genes). These results suggest that PBEF is critically involved in pulmonary vascular and epithelial inflammation and permeability, which are hallmark features in the pathogenesis of acute lung injury. This study lend further support that PBEF is a potential new target in acute lung injury.”
- eNAMPT may be the major molecular cause of normal, spontaneous labor and infection-induced pre-term labor. This was a surprising finding reported in 2002.
Reference: 2002 Pre-B-cell colony-enhancing factor, a novel cytokine of human fetal membranes
- Cancer cells secrete eNAMPT
Strong expression of eNAMPT has been seen in breast, colorectal, brain, stomach, thyroid, endometrial, ovarian, multiple myeloma, astrocytoma, and prostate cancer. This is not surprising, since cancer cells seem to up-regulate inflammatory pathways independently and autonomously from their environment. Since there is evidence that both iNAMPT and eNAMPT are pro-survival factors, there may be more than one reason why cancer cells up regulate the NAMPT gene expression. The transcription factor that upregulates eNAMPT in these cells is cMYC, an oncogene transcription factor critical in embryogenesis.
References:
Nicotinamide Phosphoribosyltransferase Promotes Epithelial-to-Mesenchymal Transition as a Soluble Factor Independent of Its Enzymatic Activity*
2011 The Role of Visfatin in Prostate Cancer
- eNAMPT triggers one of the major steps in carcinogenesis – the epithelial-to-mesenchymal transition. Cancer cells must go through a critical transformation where they go from epithelial cells to mesenchymal cells that resemble mesenchymal stem cells. This step is most often triggered by chronic inflammation and may be one reason why aspirin inhibits cancer formation. eNAMPT has been shown in vitro, to promote the epithelial-to-mesenchymal transition in breast cancer. It did this via the TGG-Beta1 signaling pathway. Thus eNAMPT plays a major role in cancer formation.
References:
Nicotinamide Phosphoribosyltransferase Promotes Epithelial-to-Mesenchymal Transition as a Soluble Factor Independent of Its Enzymatic Activity*
- eNAMPT is secreted from cardiomyocytes and causes cardiac hypertrophy and ventricular remodeling.
This is a surprising finding. NAMPT transgenic mice developed cardiac hypertrophy at 6 months of age. Cultured cardiomyocytes secreted eNAMPT in response to H2O2. The downstream signaling pathways in the cardiomyocytes that were exposed to eNAMPT were the transcription factors, JNK, p38, and ERK. There was also increased calcineurin and NFAT translocation into the nucleus in response to eNAMPT.
References:
2013 Resistin and Visfatin Expression in HCT-116 Colorectal Cancer Cell Line
- eNAMPT is a major molecular mediator of cartilage destruction in RA and OA.
This was not known until 2013. eNAMPT levels are very high in both plasma and in the synovial fluid of patients with RA. Adding eNAMPT to fibroblast cultures triggers the synthesis and secretion of cartilage breakdown enzymes like MMP-3. This effect could be reversed with the eNAMPT inhibitor, FK866.
Reference: 2013 Investigating the role of Nicotinamide phosphoribosyltransferase (NAMPT) in cartilage catabolism
- eNAMPT inhibition reduces ROS secretion by neutrophils but did not inhibit their ability to kill bacteria
Many researchers were predicting that eNAMPT would be an essential mediator of innate immunity due to its ability to activate neurotrophils. For this reason, it was somewhat surprising to find out that the inhibition of eNAMPT with FK866 decreased ROS production by neutrophils but did not reduce their ability to destroy bacteria. eNAMPT inhibition did decrease TNF-alpha mediated gene expression, however.
Reference: 2012 Regulation of neutrophil function by NAMPT
- eNAMPT inhibition by NMN prevents pro-inflammatory destruction of pancreatic islet cells.This is very good news for Type II Diabetics.
Reference: 2011 Nicotinamide mononucleotide protects against pro-inflammatory cytokine-mediated impairment of mouse islet function. This article describes a situation where administration of exogenous NMN comes to the rescue. “— We hypothesised that altered NAMPT activity might contribute to the suppression of islet function associated with inflammation, and aimed to determine whether NMN could improve cytokine-mediated islet dysfunction. — Acute effects of NMN on cytokine-mediated islet dysfunction were examined in islets incubated with TNFα and IL1β, and in mice fed a fructose-rich diet (FRD) for 16 weeks. Changes in iNAMPT, eNAMPT and inflammation levels were determined in FRD-fed mice. Results: FRD-fed mice displayed markedly lower levels of circulating eNAMPT, with impaired insulin secretion and raised islet expression of Il1b. NMN administration lowered Il1b expression and restored suppressed insulin secretion in FRD-fed mice. NMN also restored insulin secretion in islets cultured with pro-inflammatory cytokines. The changes in islet function corresponded with changes in key markers of islet function and differentiation. The anti-inflammatory effects of NMN were partially blocked by inhibition of sirtuin 1.”
- eNAMPT promotes macrophage survival via an IL-6/STAT3 survival mechanism and is differentially expressed in M1 vs M2 macrophages.
An interesting finding is that eNAMPT helps keep macrophages alive. This pro-survival effect is not due to the enzymatic activity of eNAMPT, but instead is due to the cytokine activity of eNAMPT. This pro-survival effect of eNAMPT could not be mimicked by adding NMN. It did not require the presence of Nicotinamide and it was not blocked by FK866. Thus it is clear that eNAMPT promotes survival as a cytokine in macrophages, not as an enzyme. Given the long list of “bad” things we have had to say here about eNAMPT, it is nice to identify this potentially “good” thing.
Another interesting finding is that the NAMPT gene is markedly upregulated in M1 macrophages but is down regulated in M2 macrophages. M1 macrophages are the phenotype associated with chronic inflammation whereas M2 phenotype macrophages inhibit inflammation. Exercise can phenotypically “switch” M1 macrophages to M2 macrophages, even in obese mouse models of inflammation. (See ref below). PPARgamma activation is at least one of the exercise-induced molecular mechanisms responsible for this phenotypic switch. PPARgamma is also a suppressor of inflammation and has been associated with down-regulation of eNAMPT expression.
References:
- eNAMPT makes cerebral ischemia worse. In a recent mouse model of cerebral ischemia, the non-enzymatic effect (cytokine effect) of eNAMPT was shown to exacerbate the oxygen/glucose deprivation injury. Exogenously administered eNAMPT triggered TNF-alpha release from glial cells and it appeared that this was the mechanism of injury. No enzymatic activity of eNAMPT was necessary for this effect. This eNAMPT is a “cytokine” and not an enzyme when it comes to ischemia-reperfusion injury.
References:
RECAP
In recap, here are some key facts I have covered:
- eNAMPT has a protein moiety on the protein that “mimics” endotoxin and activates the TLR4 receptor, essentially creating a “sterile inflammation.”
- eNAMPT is secreted by “angry microglial cells”, angry macrophages, and visceral fat.
- eNAMPT signaling via TLR4 triggers two pro-inflammatory intracellular cascades/transcription factors, NF-kB and AP-1. Simply inhibiting only one of these pathways like NF-kB does NOT work!
- eNAMPT expression triggers the expression and secretion of practically every SASP biomarker/cytokine, such as IL-8, IL-6, MCP-1, IL-16, TNF-a, IL-1B, CCR3, etc. Thus eNAMPT triggered secretions “mimic” cellular senescence!
- eNAMPT can be inhibited by NMN.
- eNAMPT can be inhibited by FK866 and MSO, two small molecule inhibitors of the enzyme.
- eNAMPT induces “insulin resistance” by making the IR translocated out of the lipid raft (i.e. it “pushes IR off the raft.”)
- eNAMPT induces “IGF-1 resistance” in joints by making the IGF-1R unable to trigger cell growth and cartilage ECM generation.
- eNAMPT is the “ISIS of Molecular Biology”

Hi, First, I love this blog. Next, I feel ultimately we want to break down the information into a protocol for supplementation and/or lifestyle. This is in order to optimize our health/longevity. Right? Probably you can’t recommend, but you could say something like “based on this evidence, I am cycling my XYZ supplement at 123 interval and I’m taking ABCD in this time period and EFGH in this time period.” Of course now we begin to develop a formula. Ultimately a formula may expand to include certain genetic markers. Just on this post for example, I want to boost my Curcumin. Thoughts?
Opusrun:
You are right-on in wanting to embody the tons of information in this blog and elsewhere into a protocol for lifestyle and supplementation, This has been something I have been working on for myself for over a dozen years now but is a highly dynamic challenge as new knowledge pours in at ever-increasing rates. For some of my more-recent thoughts you could check out a of my You Tube videos at
https://www.youtube.com/watch?v=dx0p7OawVcE, https://www.youtube.com/watch?v=kg1GbXk9yEk and
https://www.youtube.com/watch?v=4oe1rK6xZpA
Vince
Pingback: Inflammation Part 3: resolving inflammation – resolvins, protectins, maresins and lipoxins | AGINGSCIENCES™ – Anti-Aging Firewalls™
Pingback: ON AGING - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™
Pingback: Inflammation Part 4 - PCSK9 inhibition - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™
Pingback: Inflammation Part 2: The Tale of Three Stress Sensors and their Interactions: 1)Inflammation, 2)Genomic Instability (p53), and 3)Oxidative stress (Nrf2) - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™
Pingback: UPDATE ON LONGEVITY INTERVENTIONS – MAINLY PERSONAL - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™
Pingback: INFLAMMATION PART 6: THE SCIENCE BEHIND THE 4 HERB SYNERGY DIETARY SUPPLEMENT - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™
Pingback: My Homepage
Pingback: Inflammation Part 7: Neurohormesis, neuroinflammatory diseases, and their treatment by mushroom substances (Section 1) - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™