
By James P Watson with inputs and editorial assistance by Vince Giuliano
I. INTRODUCTION
This is the fourth in what we expect will be an open-ended series of blog posts concerned with chronic inflammation. Parts 1-6 are already published It relates to a recently discovered protein PCSK9 and tells a story of how age-related loss of LDL receptors leads to increase in LDL, associated uncontrollable cardiovascular inflammation, and atherosclerotic cardiovascular disease – the main killer of older people. It lays out why the old standard of care for atherosclerotic cardiovascular disease – the taking of statins – is slowly in the process of being augmented and replaced with a new standard of care – inhibiting PCSK9. This is also Part 1 of a new series of blog entries on interventions which are proven to increase All Cause Mortality (ACM) in large population studies. ACM statistics are the best surrogate measures we have for the actually observed longevity impacts of interventions like exercise, taking Metformin or rapamycin, or in the case of this blog entry, an intervention involving PCSK9 antibodies, or more practically supplementing with the natural phytochemical tamishoneIIA. Part 2 of this series on interventions is concermed with Metformin. Part 1 of the Inflammation series is the same as Part 5 of the NAD world. That blog entry is concerned with The pro-inflammatory effects of eNAMPT(extracellular NAMPT, nicotinamide phosphoribosyltransferase). Part 2 of the Inflammation series relates a) the “master” pathway network of inflammation (NF-kB) to two other pathway networks clearly implicated in aging and disease processes, b) Genomic Instability (p53), and c) Oxidative stress (Nrf2). Part 3 of the Inflammation series of blog entries is concerned with the all-important resolution phase of inflammation, how acute inflammation goes away under ideal conditions instead of hunkering down to lingering and dangerous chronic inflammation. It is concerned with recently identified substances found in fish and flaxseed oils that play important roles in resolving certain kinds of inflammation – what they can do and how they work. Part 5 of the series is concerned with the basic science of inflammasomes and how they relate to a number of disease processes. Part 6 of this series is concerned with the scientific considerations behind a dietary substance I have invented to control chronic inflammation.
Atherosclerotic cardiovascular disease is the leading cause of morbidity and mortality worldwide (Gupta, Expert Review, Dove Press). Wannah die later rather than sooner? Then pay attention to this.
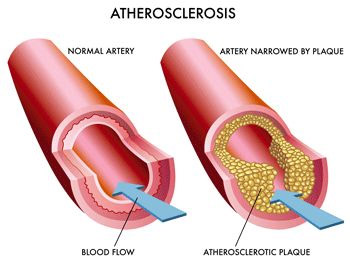
Many National and International though leaders concerned with the disease have refocused their efforts from lowering cholesterol to lowering LDL levels (Grundy, et.al., 3rd Report of NECP). The reason for this is due in part to the discovery of PCSK9 protein, which is a circulating protein in blood that triggers the degradation of the LDL-receptor.
First, a little history about the discovery of the PCSK9 gene and why all the excitememnt about it. PCSK9 is an abbreviation for Preprotein Convertase Subtilisin/kexin type 9, an extracellular protein that triggers the degradation of the LDL receptor, the LRP-1 receptor, and other receptors found on the surface of many cells (especially the liver). In 2003, French researchers reported two mutations in the PCSK9 gene that caused familial autosomal dominant hypercholesterolemia (Abifadel, et.al., Nature Genetics). In 2008, Spanish researchers found a new mutation in the promoter of the PCSK9 gene that increased gene expression of PCSK9 mRNA and plasma levels of PCSK9 (Blesa, et.al. J Clin Endocrin Met). About the same time, researchers at UT Southwestern in Dallas reported a series of PCSK9 gene nonsense mutations found in 2.6% of a large group of African Americans that reduced LDL plasma levels by 15% and reduced coronary artery disease risk by 47% (Cohen, et.al., NEJM). In the ensuing 10 years, monoclonal antibodies that target the circulating PCSK9 protein have been developed, tested in over 25 R clinicaltrials, and FDA-approved.
What these clinical trials have shown in their “size effect” of reducing all-cause mortality (ACM) is nothing short of astounding – much more than statins ever did. All cause mortality is reduced 55% in patients with hypercholesterolaemia. So, we go on now to discussing how this all works.
II. MECHANISMS RELATING LDL, ATHEROSCLEROSIS AND HEART DISEASE, LDL CELL RECEPTORS, PCSK9 AND MOST OF THE MOLECULAR USUAL SUSPECTS OF INFLAMMATION, DISEASE AND AGING
It has long been known that there is a positive correlation between circulating LDL, atherosclerosis and heart disease. Why is this? The most harmful aspect of high LDL levels is most likely due to the fact that LDL is oxidized to “oxidized-LDL” (oxLDL) by free radicals (ROS) which triggers a pro-inflammatory receptor found on vascular endothelial cells called the “Lectin-like oxidized low density lipoprotein receptor 1” (LOX-1). Here is a diagram of how this works:
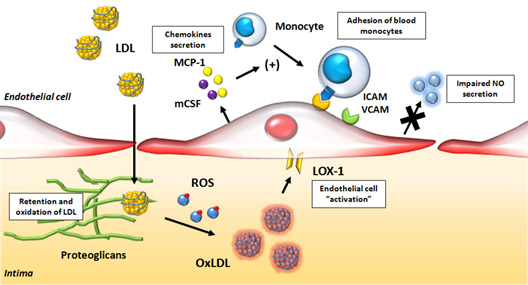
Image source and reference: Role of Oxidized LDL in Atherosclerosis
Oxidized LDL lipoproteins bind to the LOX-1 receptor and are then internalized into endothelial cells, vascular smooth muscle cells, as well as monocyte/macrophages. Inside the cell, this process triggers free radical production (ROS) and activation of NF-kB (the master switch for inflammation). NF-kB then turns on the transcription of hundreds of inflammatory genes, including cytokines (IL-8) and chemokines (CXCL2, CXCL3, CSF3), which are then secreted and trigger the CXCR2 receptor on white blood cells. This is how the inflammatory cascade is “let loose” when your LDL-C is too high. Here is a further diagram of how this works:
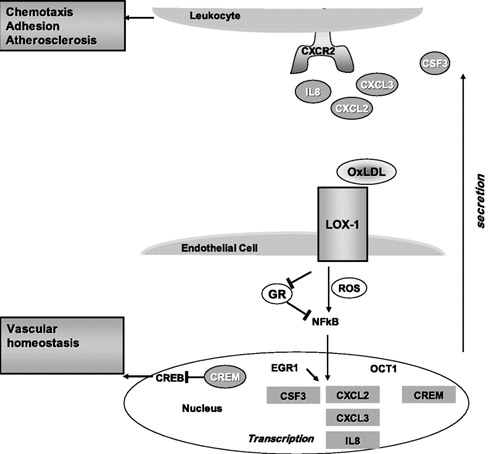
Diagram and legend source: LOX-1-dependent transcriptional regulation in response to oxidized LDL treatment of human aortic endothelial cells (2009) “Model for LOX-1 functions in atherosclerosis and endothelial dysfunction. LOX-1 binding to OxLDL initiates reactive oxygen species (ROS) formation and an inflammatory response mediated in part by nuclear factor (NF)-κB and EGR1, leading to an upregulation and secretion of chemokines. The three CXC-chemokines (CXCL2, CXCL3, and IL-8) are all ligands for the same receptor, CXCR2, found on leukocytes. CXCR2 activation leads to chemotaxis and adhesion of leukocytes to endothelium. EGR1, CXCR2, and CSF3 are all implicated in animal models of atherosclerosis and represent novel molecular connections between LOX-1 and atherosclerosis. The transcription factor CREB is involved in maintaining vascular homeostasis. LOX-1 activation by OxLDL leads to a downregulation of CREB target genes and an upregulation of the CREB repressor CREM, thus providing a potential molecular mechanism of LOX-1-dependent endothelial dysfunction.”
In addition to LOX-1 mediated vascular inflammation, oxLDL/LOX-1 internalization triggers endothelial/vascular smooth muscle cell dysfunction, apoptosis, cellular senescence, or osteoblastic differentiation. The osteoblastic differentiation of these cells is manifested as “vascular calcification”. If the oxLDL/LOX-1 internalization occurs in a monocyte, the macrophage is phenotypically transformed into the classic “foam cell”, which is the sine qua non of atherosclerosis. Along with the other 4 “drivers” of atherosclerosis (Angiotensin II, pro-inflammatory cytokines, sheer stress, and advanced glycation end products) this oxLDL/LOX-1 pathway is considered to be the major molecular cause of the disease.
So, the first point in the story is this: 1. Too high LDL is bad because when oxidized it lets loose an inflammatory cascade that can lead to chemotaxis, adhesion, plugged arterial lumens, atherosclerosis and heart disease.
The next part of the story is simple: 2. under normal healthy conditions, LDL receptors on cells latch on to LDL particles and drag them into the cell where endosomes degrade the LDL and then the receptors pop back up to the surface of the cell and wait for another LDL particle to come along. But if PCSK9 is present, the LDL receptor itself is destroyed. The amazing clinical results mentioned above were due to inhibiting PCSK9.
The diagram below illustrates how PCSK9 works for elimination of LDL receptors and therefore reduction of capability to eliminate LDL.
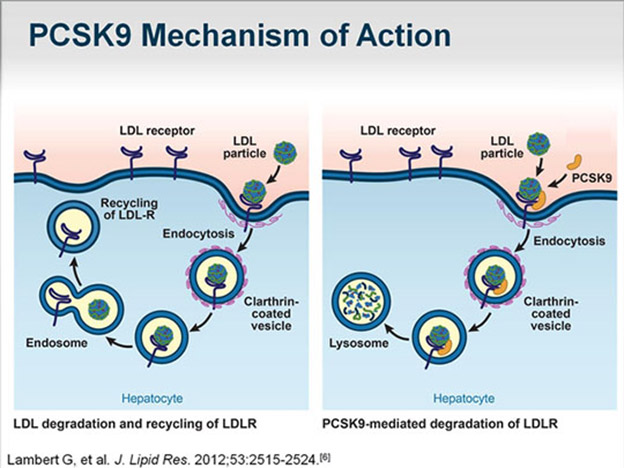
Image source: Complementing the “Gold Standard”: Exploring PCSK9 MOA with Current Lipid Therapies “When PCSK9 is present in that complex, what ultimately happens is that the LDL receptor is subsequently targeted via the endosome to the lysosome for degradation (situation depicted in right panel in diagram). Hence, we lose the LDL receptor; it does not recirculate back to the liver. If there is no PCSK9 that is bound to that complex, then once it is internalized, the cholesterol is released, the LDL particle is released from the receptor, and the receptor recirculates back to the surface, where it can then attach to more LDL cholesterol and clear more LDL cholesterol from the circulation degradation (situation depicted in left panel in diagram).”
The following diagram show how the final part of the story works: 3. anti-PCSK9 antibodies prevent PCSK9 from binding to LDL receptors so they are not destroyed in the lysosome.
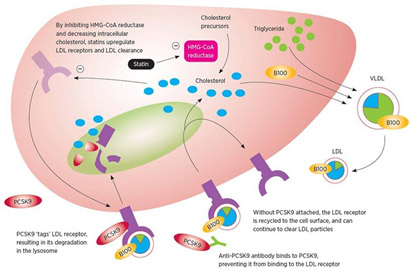
DIAGRAM B Image source
As already mentioned, although the PCSK9 inhibitor class of drugs have only been recently FDA approved, they have already shown the largest ACMR (all-cause mortality reduction) of any class of FDA-approved drugs ever approved in history.
III. PCSK9 AND AGING
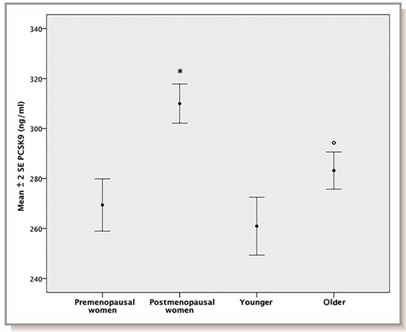
These drugs are especially important for those of us who are interested in the science of aging since it has been shown that the levels of PCSK9 protein circulating in the plasma increases with aging in rodents and in humans (Tao, et.al., J Bio Chem) (Ruscica, et.al. J Am Heart Asso).
Here is a diagram showing the relative levels of plasma PCSK9 in pre vs post menopausal women and young vs old men in a large population-based study (Diagram source: Ruscica, et.al. J Am Heart Asso).
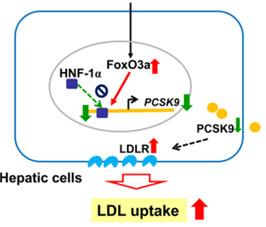
The cause of the age-related increase in PCSK9 gene expression is due to a loss of suppression of the PCSK9 gene by the FoxO3a transcription factor. Normally SIRT6 recruits FoxO3a to the promoter of the PCSK9 gene, but with aging, SIRT6 activity declines due to declining NAD+ levels in the cell. As a result, there is an increase in PCSK9 gene expression with aging (Tao, et.al., J Biol Chem). Here is a diagram illustrating this:
![]()
Note that we are here extending the story line of our NAD World blog entries detailing negative impacts of declining NAD+ LEVELS.
Reference and image source :Role of Oxidized LDL in Atherosclerosis (2015)
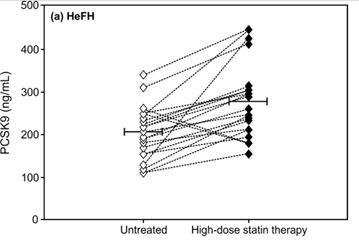
FoxO3a and Nrf2 are considered two of the most important transcription factors that protect mammals from aging by increasing oxidative stress resistance (Li, et.al. Ox Med Cell Long). Multiple genetic variants in the Foxo3a gene have been linked in many studies world-wide with extreme longevity (Anselmi, et.al., Rejuvenation Research). SIRT6 has also been shown to be a longevity gene in GWAS studies as well as in lab animals (Braidy, et.al., Front Cell Neuroscience). Thus it is not surprising that aging due to declines in FoxO3a/SIRT6 activity triggerw the increase in expression of the PCSK9 gene and play a major role in vascular aging. Moreoever, statin use causes a paraxodical INCREASE in PCSK9 levels in patients with familial hypercholesterolemia, most likely due to a compensatory homeostatic feedback mechanism (Raal, et.al., J Am Heart Asso). The following diagram is from this study.
Another reason why PCSK9 levels increase with aging is insulin signaling. It is not surprising either that Insulin/IGF-1 signaling exacerbates this problem with age-related insulin resistance, since FoxO3a cannot enter the cell nucleus under conditions of high glucose/insulin signaling (due to the inhibition of nuclear translocation of FoxO3a by Akt). This is why diabetes, metabolic syndrome, and insulin resistance leads to an increase in the expression of PCSK9 gene. Here is a diagram of the relationship between insulin resistance (HOMA-IR) and insulin correlated with increases in PCSK9 expression over time
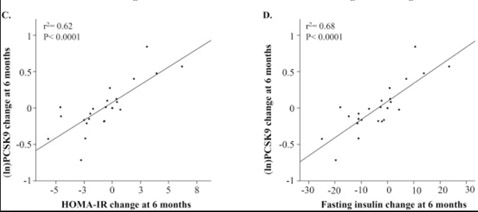
(Image source: Levenson, et.al. NMCD).
Age and diet-related increases in LDL blood levels are probably the most common clinical problem facing modern man. Although statins reduce endogenous cholesterol synthesis, they do not directly stop the age-related increase in PCSK9 protein in the blood. As a result, most studies have suggested that “age” is the greatest risk factor for atherosclerosis after age 50, whereas “cholesterol” is the greatest risk factor for atherosclerosis prior to age 50. For this reason, the value of a drug that reduces the PCSK9 protein in the blood should increase with aging. So far, this prediction has held true. Unlike statins, where their value declines in old age (zero value by age 85), there appears to be value in reducing LDL levels even in old age, with FoxO3a and SIRT6 signaling decline.
So, in this overall story we are seeing a link-up of several “usual suspect” themes we have touched on multiple times before in this blog: insufficient expression of sirtuins and lack of NAD+, the FoxO3a transcription factor, high age-related LDL, the Insulin/IGF-1/PI3K/Akt pathway, the master trigger of inflammation NF-kB, Nrf2, reactive oxygen species, inflammatory cytokines, atherosclerosis. With a new element which is age-related overexpression of the PCSK9 gene which kills off LDL receptors.
Cholesterol, step to the rear of the coach please
Whereas cardiologists used to consider cholesterol on this “big five” list, it is no longer considered to be as important as it once was. Instead, high blood levels of LDL is now reputed to be the #1 culprit that causes atherosclerotic disease. Since PCSK9 proteins trigger the degradation of the LDL receptor, monoclonal antibodies against PCSK9 have been successful at dropping LDL levels as low as 10 mg/dl (although no physician is recommending that you lower your LDL that much). Getting rid of LDL reduces oxidized LDL, which dramatically reduces LOX-1 activation. Reduced LOX-1 activation reduces endothelial cell dysfunction, apoptosis, senescence, and osteoblastic differentiation of endothelial cells (which is a major cause of vascular calcification). It is this mechanisms that explains why PCSK9 inhibitor therapy has been shown to reduce vascular and valvular calcification (something that statins rarely do).
Despite these known (theoretical) molecular mechanisms, most cardiologists were skeptical that PCSK9 inhibitors would reduce all-cause mortality as well as statins. However, the results of the PCSK9 inhibitor clinical trials have shattered all doubts by 10 miles! The results on ACM reduction have been nothing short of amazing. A meta-analysis of the 24 RCTs that have been done so far (N = 10,159) show a 55% ACMR (OR = 0.45) and a 50% reduction in cardiovascular mortality (OR = 0.50). The rate of MI was also reduced by 51% (OR = 0.49). Even increases in serum creatinine kinase (CPK) was reduced (OR = 0.72). Because PCSK9 levels increase as a function of aging, it is likely that these drugs will still have beneficial effects after age 85, whereas data shows that statins probably become harmful after age 85.
F In addition to LOX-1 mediated vascular inflammation, oxLDL/LOX-1 internalization triggers endothelial/vascular smooth muscle cell dysfunction, apoptosis, cellular senescence, or osteoblastic differentiation. The osteoblastic differentiation of these cells is manifested as “vascular calcification”. If the oxLDL/LOX-1 internalization occurs in a monocyte, the macrophage is phenotypically transformed into the classic “foam cell”, which is the sine qua non of atherosclerosis. Along with the other 4 “drivers” of atherosclerosis (Angiotensin II, pro-inflammatory cytokines, sheer stress, and advanced glycation end products) this oxLDL/LOX-1 pathway is considered to be the major molecular cause of the disease.
DIAGRAM A above, shows how PCSK9 inhibitors work and how they are fundamentally different than statins:
Reference: Effects of Proprotein Convertase Subtilisin/Kexin Type 9 Antibodies in Adults With Hypercholesterolemia: A Systematic Review and Meta-analysis (2015)
IV. TURNING TO THE PERSONAL AND PRACTICAL
Costs of PCSK9 Inhibitor Therapy and Delivery methods – $14,000/year in the US but cheaper overseas
This blog is designed to be a practical resource that you can use on your personal journey to improved healthspan. It is not a substitute for your personal physician, however. For this reason, we suggest you consult with your doctor before attempting any of the interventions in this blog, including monoclonal antibodies against PCSK9. However, your doctor may be completely unaware of this new class of drugs and your health insurance company will not pay for the drugs unless you have had an MI or a stroke AND you also have failed to control your LDL with statins (or have severe side effects from statins that cannot be treated with CoQ10). In the US, the annual costs for PCSK9 inhibitor therapy runs about $14,000 per year so this is mow a very expensive way to try to lengthen your healthspan. However the cost of the same PCSK9 inhibitor drug in some other countries can be half the price found here in the US, so for those who have figured out how to do cash-based medical tourism, you can save $7,000 per year. Here are the two monoclonal antibodies against PCSK9 that are currently FDA-approved:
- Alirocumab (Praluent) – This monoclonal antibody against the PCSK9 protein was the first to be approved by the US FDA (July, 2015). It is a twice-a-month SQ injection of 75-150 mg per dose. It has been FDA-approved for patients with heterozygous familial hypercholesterolemia or for normal patients whose cholesterol cannot be adequately controlled with diet and statins. Patients who go on Praluent are supposed to stay on their statin, even though they may have to lower their dose to avoid statin-induced muscle pain. Side effects include nasopharyngitis (11%), injection site reactions (7%), influenza (5.7%), UTIs (4.8%), and diarrhea (4.7%). Neurocognitive events were seen in two of the large clinical trials, but did not correlate with how low the LDL-C went. Further studies have failed to show any neurocognitive difference between the drug and the placebo group (0.7-0.8%). 8% of patients developed anti-drug antibodies. Alirocumab costs $1200 per month or $14,350 per year in the US. What is amazing is that PCSK9 monoclonal antibody therapy can drop your LDL-C to as low as 25 with no evidence of any major side effect. With such dramatic reduction, vascular and valvular calcifications have been shown to be reduced.
- Evolocumab (Repatha) – This monoclonal antibody was approved in August, 2015, shortly after Alirocumab. Like Alirocumab, it is approved for familial (genetic) hypercholesterolemia (heterozygous or homozygous) and patients who fail diet + statin therapy. Unlike Praluent, it can be given twice-a-month dose (140 mg) or once-a-month (420 mg) SQ. Like Alirocumab, patients are supposed to remain on statins. Most common side effects Include nasopharyngitis (11%), URI (9.3%), influenza (7.5%), back pain (6.2%), and Injection-site reactions (5.7%). Ongoing studies are closely looking for neurocognitive side effects, but these studies are not completed. The annual costs for Evolocumab are almost identical to those for Alirocumab.
Are PCSK9 Inhibitors Cost Effective? – Answer: No
While our enthusiasm for this dramatic reduction in all-cause mortality may appear to be “exuberant”, a famous economist once said that people often suffer from “irrational exuberance”. In the case of PCSK9 monoclonal antibodies, this is definitely true. The “bean counters” have already figured out that these new drugs are overpriced to be cost effective (Really!….any 5th grader could tell you that!). At $14,000 per person per year, the two PCSK9 inhibitor drugs are expected to generate $4.5 billion USD in annual sales by 2020 (Big Pharma is drowning in their saliva over this!). If all eligible patients took these injections, it would add an approximately $150 billion annual cost to our health care system. Only if the price of these drugs dropped to $4,536 per patient per year would the drugs start to become cost effective. However. if you have a high LDL-C despite eating a strict cholesterol-free diet and taking your statins, taking one of these drugs could lengthen your healthspan.
Reference: Are PCSK9 meds worth the cost? Only if Amgen, Sanofi and Regeneron slash prices by two-thirds: JAMA
Alternative to PSCK9 Inhibitor Therapy – Reduce Insulin signaling with diet, SIRT6 activation, fasting, and the Salvia extract, Tanshinone IIA
For those who do not have $14,000 per year to “burn”, there is a much simpler way of “shutting off” PCSK9. As mentioned in the beginning of this section, the “longevity gene” FoxO3a functions as a suppressor of PCSK9 gene expression when the FoxO3a transcription factor can enter the cell nucleus. Unfortunately to do this, you must reduce insulin signaling, since insulin prevents FoxO3a from entering the cell nucleus (due to the Insulin/IGF-1/PI3K/Akt pathway). Once you reduce insulin signaling, FoxO3a can enter the cell nucleus and be recruited by SIRT6.
Unfortunately, with aging, SIRT6 cannot be activated due to declining levels of NAD+ within the cell. This can be ameliorated (at least in theory) by restoring NAD+ levels within the cell with NAD+ precursors like NR or NMN, or possibly with NAD+ given IV. (you can look at our blog postings on the NAD World that describe the mechanisms involved.) Specifically, however there is a fascinating phytochemical that has been isolated from the Salvia miltiorrhiza Bunge plant called Tanshinone IIA. This compound is the most pharmacologically bioactive compound found in the Salvia plan and has anti-inflammation, anti-cancer, anti-LDL-cholesterol, neuroprotective, and hypolipidemic properties. Only recently was the molecular mechanism of the Tanshinone IIA compound elucidated by a team of researchers from Taiwan. Below is a molecular diagram of the molecule and the reference for how it works.
There are other molecular mechanisms that regulate the PCSK9 gene besides the SIRT6-mediated FoxO3a suppression, however. This includes binding sites for several transcription factors in the promoter region of the PCSK9 gene (SREBP-1/2, HNF1A, farnesoid X receptor, PPAR-gamma, liver X receptor, and histone nuclear factor P). Fasting has also been shown to be a very powerful way of reducing plasma levels of PCSK9, reducing PCSK9 levels by as much as 58%, compared to fed conditions. This is an even greater effect than the monoclonal antibodies!
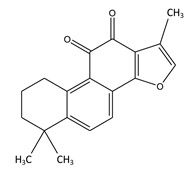
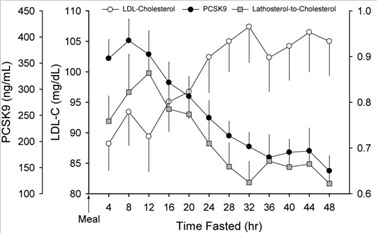
Image and legend source Effects of fasting on PCSK9 levels “Trends in plasma LDL-c, PCSK9, and lathosterol-to-cholesterol ratio during a 48 h fast. Plasma levels of LDL-c increased modestly over the initial 32 h of the two-day fast (P = 0.004) and then stabilized. Plasma PCSK9 levels declined steadily after 8 h of fasting and reached a nadir by 36 h (P = 0.024). The decline in plasma PCSK9 occurred in tandem with a decrease in the lathosterol-to-cholesterol ratio (P = 0.091). Data (mean ± SEM) were derived from 18 healthy subjects (described in Table 1) who underwent an observed 48 h fast. PCSK9, proprotein convertase, subtilisin/kexin type 9; LDL-c, low-density lipoprotein cholesterol.”
References:
- Tanshinone IIA Modulates Low Density Lipoprotein Uptake via Down-Regulation of PCSK9 Gene Expression in HepG2 Cells (2016)
- Anti-Inflammatory Activity of Tanshinone IIA in LPS-Stimulated RAW264.7 Macrophages via miRNAs and TLR4-NF-κB Pathway (2016).
- Fasting reduces plasma proprotein convertase, subtilisin/kexin type 9 and cholesterol biosynthesis in humans (2010)
Conclusion: The story of the strange, extracellular protein called PCSK9 has now become the focal point of a $4.5 billion/year gravy train for Big Pharma companies, due to the dramatic effect that monoclonal antibodies have of clearing this “bad” protein out of the blood. There is no debate that the use of these monoclonal antibodies has a greater effect on reducing all-cause mortality than any other drug, diet, supplement, or lifestyle intervention at this time, with an ACM reduction of 55%. However until the cost of these PCSK9 monoclonal antibodies drops below $4,536 per person per year, they are not cost effective. Since the increased expression of the PCSK9 gene is due in part to the Insulin/IGF-1 signaling pathway, which prevents FoxO3a transcription factor from migrating into the cell nucleus, the most cost effective way to reduce PCSK9 expression is to reduce your insulin levels. This can easily be done with diet and exercise. Moreover, a phytochemical from the Salvia plant called Tanshinone IIA, and SIRT6 activation with NAD+/NR/NMN may also be ways to prevent the increase in PCSK9 gene expression which are NOT due to insulin. The loss of SIRT6 activity due to the decline in NAD+ levels within the nucleus is a likely contributor to the age-related increase in PCSK9. Nevertheless, because oxidized LDL is such a powerful “driver” of atherosclerosis, a dramatic reduction in PCSK9 plasma protein levels should be a major goal for lengthening lifespan. Despite its unpopularity, fasting has been scientifically shown to be the cheapest and most effective way to reduce PCSK9. With a 48-hour fast, PCSK9 levels can be lowered by 58%, which is even more than the monoclonal antibodies do!
We have additional blog entries in both the inflammation series and the ACM intervention series in draft form and expect that some of these will be published soon.
MEDICAL DISCLAIMER
FROM TIME TO TIME, THIS BLOG DISCUSSES DISEASE PROCESSES. THE INTENTION OF THOSE DISCUSSIONS IS TO CONVEY CURRENT RESEARCH FINDINGS AND OPINIONS, NOT TO GIVE MEDICAL ADVICE. THE INFORMATION IN POSTS IN THIS BLOG IS NOT A SUBSTITUTE FOR A LICENSED PHYSICIAN’S MEDICAL ADVICE. IF ANY ADVICE, OPINIONS, OR INSTRUCTIONS HEREIN CONFLICT WITH THAT OF A TREATING LICENSED PHYSICIAN, DEFER TO THE OPINION OF THE PHYSICIAN. THIS INFORMATION IS INTENDED FOR PEOPLE IN GOOD HEALTH. IT IS THE READER’S RESPONSIBILITY TO KNOW HIS OR HER MEDICAL HISTORY AND ENSURE THAT ACTIONS OR SUPPLEMENTS HE OR SHE TAKES DO NOT CREATE AN ADVERSE REACTION.

{kind=link}
{kind=link}
Pingback: Part 5 of the NAD world: the conflicting roles of NAMPT – inflammation or rescue? Also Part 1 on a new series on inflammation - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™
By fasting do you mean water only? How frequently? That was another great article!
Pingback: Part 2 of interventions that reduce all cause mortality (ACM) - Metformin - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™
Pingback: Inflammation Part 7: Neurohormesis, neuroinflammatory diseases, and their treatment by mushroom substances (Section 1) - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™