
By James P. Watson and Vince Giuliano
Inflammasomes are multimeric self-assembling protein complexes within the cytosol of mammalian cells, pattern-recognizing components of the innate immune system. They can be thought of as finely tuned alarm, triggering and amplifying systems that are responsive to cellular stresses and infections. When activated they can promote the maturation and release of pro-inflammatory cytokines and danger signals as well as pyroptosis, a rapid, pro-inflammatory form of cell death.
Inflammasomes are among the newest (discovered in 2002), most complex, and least understood entities encountered related to inflammation,. They are very interesting because a) their actions can explain phenomena that were previously completely enigmatic such as sterile inflammation and the actions of beta amloid and tau tangles in Alzheimer’s disease. b) Inflammasomes play important roles in a number of chronic disease processes. And c) practical actions can be taken to control their activation and consequently limit runaway chronic inflammation. It appears that, certain plant based substances, for example reseveratrol, can be effective in this regard.
This is Part 5 of what will likely be a nine-part series of blog entries on inflammation. Being a central aspect of every degenerative disease of old age, chronic inflammation can be thought of as the Great Executioner, the most central machinery of most people’s ultimate illnesses and deaths. This blog entry is concerned with the basic science of inflammasomes and how they relate to a number of disease processes. There is much to be said also about how practically to limit or prevent the formation or expression of inflammasomes, or otherwise impact them. We are planning to discuss them in a Part 6 of this series on inflammation which will be on Substances That Can Limit or Prevent The Formation or Expression of Inflammasomes.
Parts 1 through 6 of this series related t0 chr9nic inflammation are already published. Part 1 of the series is the same as Part 5 of the NAD world. That blog entry is concerned with The pro-inflammatory effects of eNAMPT(extracellular NAMPT, nicotinamide phosphoribosyltransferase). Part 2 relates 1) the “master” pathway network of inflammation (NF-kB) to two other pathway networks clearly implicated in aging and disease processes, 2) Genomic Instability (p53), and 3) Oxidative stress (Nrf2). Part 3 is concerned with the all-important resolution phase of inflammation, how acute inflammation goes away under ideal conditions instead of hunkering down to lingering and dangerous chronic inflammation. Part 4 of the inflammation series, is concerned with PCSK9 inhibition – Also that blog entry is Part 1 of a series on interventions that reduce all cause mortality (ACM). Part 5 of the series is concerned with the basic science of inflammasomes and how they relate to a number of disease processes. Part 6 of this series is concerned with the scientific considerations behind a dietary substance I have invented to control chronic inflammation.
A closely related blog entry The Making of a Dietary Supplement is the story of how that dietary supplement was conceived, invented and tested to control chronic inflammation and tested by my colleagues and I. This supplement is made of synergistic combination of traditional anti-inflammatory herbs and offers unusual bioavailability because of its liposomal nature. Specifically, the blog entry is about the scientific basis for the supplement.
An anticipated, this inflammation-related blog series is likely to be open-ended. For example, a future part will likely be concerned with the frequent decoupling of transcription and translation in protein synthesis, the code for what goes on at the Internal Ribosome Entry Sites” (or IRES). And it will be concerned with implications of decoupling for both generation of inflammation and its consequences, particularly insulin resistance. Additional parts will likely be concerned with certain other traditional herbal remedies used for control of inflammation, such as Lion’s Mane mushroom extract. And as new research emerges I will continue to report on how the effectiveness and bioavailability of some such herbs can be multiplied by judicious combination and by current nano-delivery techniques as ancient Chinese and Ayurvedic medicine joins up with high-tech in the emerging present.
Some bottom-line messages of this blog entry
As seen by Vince
- Inflammasomes have existed in mammalian species for hundreds of millions of years. They are key parts of our inane immune defense system. They play key roles in activating inflammation and assuring its continuance as long as it is needed, and protecting us against a large variety of assaults that would otherwise be deadly, including viral assaults and toxins.
- But inflammasomes have been recognized in science only since 2002 so there is much still to be learned about them. And they are hardly recognized or acknowledged yet for their potential importance in medical practice.
- Inflammasomes are necessary and useful, and they are also part of the programmed pruning apparatus that kills off older people. They are also closely associated with the initiation and progression of many diseases.
- What we know about inflammasomes is consistent with most if not all the previous knowledge we have accumulated about inflammation and inflammatory processes, and what can be done about them.
- Yet, embracing inflammasomes has already extended our understanding in very basic ways. They help explain chronic inflammation, age-related inflammation, and inflammatory diseases for which there are no inflammatory pathogens, like Alzheimer’s and Parkinson’s Diseases. And they provide new more-satisfying explanations for why some time-honored approaches to controlling inflammation work and others do not.
- Reviewing what we know now about inflammasomes, we can say their story is a major and essential chunk of the inflammation story, a chunk that we must grapple with if we are to understand chronic inflammation and best bring it under effective control.
- Understanding inflammasomes may be as important a step for dealing with chronic inflammation as understanding microbes once was for dealing with infectious diseases.
- The good news is that inflammasomes can be hacked. Old-friend phytosubstances like resveratrol, quercetin and curcumin can inhibit cytokine release from them (the activation phase), and a number of others can inhibit their formation in the first place (the priming phase). We expect to deal with such hacks in our expected Part 6 blog entry.
OVERVIEW ON INFLAMMASOMES
An inflammasome is a cytosolic high molecular weight protein complex that can detect and be activated by any of a number of signal sources that could initiate a possible problem for the cell, including microbial toxins, live bacteria, xeno-compounds, viruses, fungi, mycobacteria, or Protozoa exposure. These activators are also known as Pathogen-Associated Molecular Patterns (PAMPs) and Danger-Associated Molecular Patterns (DAMPs). A number of different inflammasomes have been identified including a number named because of the NLR protein they contain, including NLRP1, and the most-studied one NLRP3. Others include the IPAF (NLRC4) inflammasome. Not all inflammasomes contain a member of the NLR family, such as the recently-discovered AIM2 (absent in melanoma) inflammasome. The primary purpose of the Inflammasome is to trigger the activation of the mature form of Caspace-1, IL-1 and IL-18, potent inflammatory cytokines that are secreted by the cell after activation by the Inflammasome. Another purpose can be to accelerate cell death via pyroptosis. The Inflammasome components are mostly proteins encoded by NF-kB transcription factor regulated genes. However the JAK-STAT pathway also plays a role.
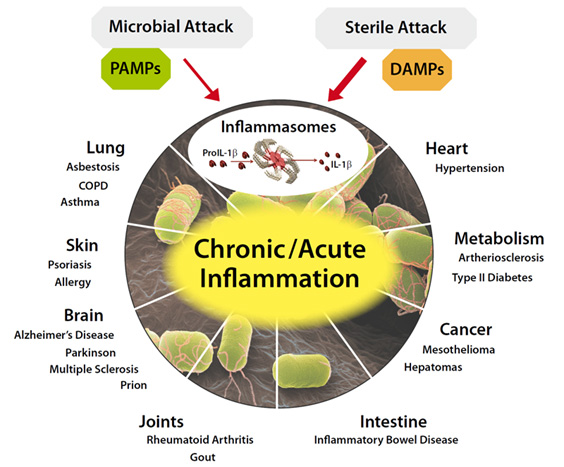
Image source and further introduction to inflammasomes
A good review article on inflammasomes is NLRP3 inflammasome and its inhibitors: a review. “Inflammasomes are newly recognized, vital players in innate immunity. The best characterized is the NLRP3 inflammasome, so-called because the NLRP3 protein in the complex belongs to the family of nucleotide-binding and oligomerization domain-like receptors (NLRs) and is also known as “pyrin domain-containing protein 3”. The NLRP3 inflammasome is associated with onset and progression of various diseases, including metabolic disorders, multiple sclerosis, inflammatory bowel disease, cryopyrin-associated periodic fever syndrome, as well as other auto-immune and auto-inflammatory diseases. Several NLRP3 inflammasome inhibitors have been discovered. There is so great chemical diversity among 2nd-phase inflammasome activators, that it is thought that they may induce a common cellular stress response instead of binding directly to the NLRP3 inflammasome.”
A very interesting property of inflammasomes is that the formation self-assembly phase (priming phase) is decoupled from the activation phase, both of which happen in response to different stimuli. Once formed, inflammasomes may lurk for long periods and be responsible for persistent seemingly-uncaused (“sterile”) chronic inflammation. This is the kind of inflammation associated with advanced aging and the chronic diseases of aging that kill older people.
The assembly phase is described in the article Structural mechanisms of inflammasome assembly. (2015) ”Inflammasomes are supramolecular signaling complexes that activate a subset of caspases known as the inflammatory caspases, an example of which is caspase 1. Upon stimulation by microbial and damage-associated signals, inflammasomes assemble to elicit the first line of host defense via the proteolytic maturation of cytokines interleukin-1β and interleukin-18, and by induction of pyroptotic cell death. Inflammasome assembly requires activation of an upstream sensor, a downstream effector and, in most cases, an adaptor molecule such as apoptosis-associate speck-like protein containing a caspase recruitment domain (ASC). Depending on whether ASC is required, inflammasomes can be categorized into ASC-dependent and ASC-independent inflammasomes. — Canonical inflammasomes assemble upon the activation of two classes of sensor molecules: NOD-like receptors (NLRs) and AIM2-like receptors (ALRs). The human genome encodes 22 NLRs [3], but only NLRP1 [4], NLRP3 [5, 6], NLRP6 [7], NLRP7 [8], NLRP12 [9] and the NAIP/NLRC4 complex [10, 11] have been reported to assemble into their respective inflammasomes. These essential sensor proteins are composed of an effector domain such as the caspase recruitment domain (CARD), Pyrin domain (PYD) or baculoviral IAP repeat domain for downstream interactions, a nucleotide-binding and oligomerization domain NACHT, and a leucine-rich repeats domain (Fig. 1).”
This diagram offers another view of inflammasome assembly:
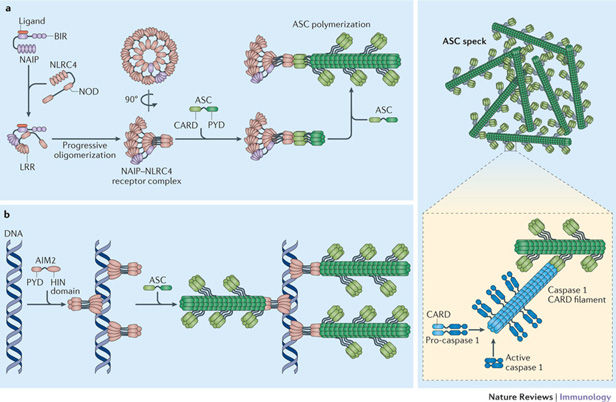
Image and legend from Inflammasomes: mechanism of assembly, regulation and signalling (2016) Formation of The NAIP–NLRC4 inflammasome. “Binding of their specific ligand activates mouse and human NLR family, apoptosis inhibitory proteins (NAIPs) and allows them to interact with and activate NLRC4 (Refs 21,22,23). This interaction results in the generation of a new nucleating surface, which in turn allows NLRC4 molecules to recruit and further activate NLRC4 molecules in a domino-like reaction68, 69. The completed NAIP–NLRC4 complex is a multi-subunit disk-like structure that contains 9–11 molecules of NLRC4, but only one NAIP molecule68, 69. Following receptor complex assembly, the caspase recruitment domains (CARDs) of NLRC4 cluster and recruit bridging ASC molecules to initiate ASC polymerization. A NAIP–NLRC4 complex can also directly initiate caspase 1 activation in the absence of ASC (not shown), although ASC recruitment seems to be the default situation. b | The absent in melanoma 2 (AIM2) inflammasome. DNA is bound in regular intervals by the HIN domain of AIM2, resulting in the clustering of the AIM2 pyrin domains (PYDs)75, 76. This allows the PYDs of AIM2 to recruit ASC and to initiate ASC polymerization into filaments. ASC filaments aggregate to form a macromolecular assembly that is known as the ASC speck. The CARD of ASC, which is exposed on the surface of the ASC filaments recruits pro-caspase 1 and in turn initiates filaments that are formed by the CARD of caspase 1 and that are necessary for proximity-induced activation of the caspase79, 80. LRR, leucine-rich repeat; NOD, nucleotide-binding oligomerization domain.”
A central message portrayed in the following diagram is that the roles of the different inflammmasomes are to protect us from assaults that could otherwise be deadly: lethal toxins in the case of NLRP1. PAMPS and DAMPS in the case of NLRP3, Salmonella in the case of NLRC4, F novicida in the case of AIM2, and some toxins in the case of Pyrin (as examples).
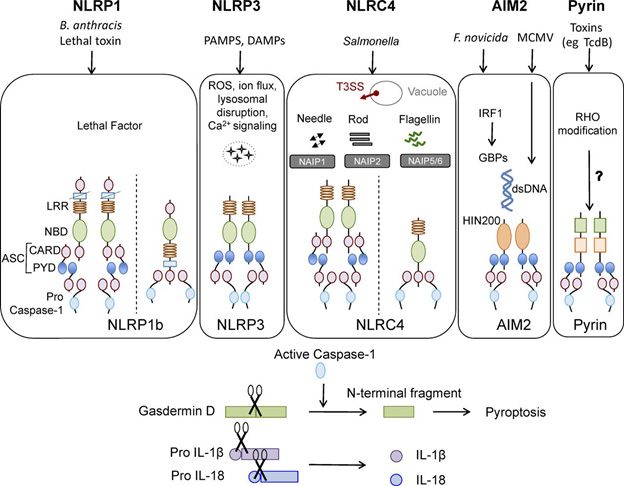
Some canonical inflammasomes and factors influencing their activation.
Image and legend source: 2016 review The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. “Canonical inflammasomes NLRP1, NLRP3, NLRC4, AIM2, and pyrin. Ligands and upstream mediators involved in inflammasome activation. The NLRP1b inflammasome responds to lethal factor produced by B. anthracis and assembles an inflammasome by recruiting caspase-1 through its CARD domain or through ASC as an adaptor. The NLRP3 inflammasome responds to intracellular damage induced by pathogenic or sterile insults. The NLRC4 inflammasome assembles in response to recognition of bacterial flagellin or components of the type III secretion system via NAIPs and can form complexes with or without recruiting ASC. AIM2 inflammasome senses double-stranded DNA through its HIN200 domain. Pyrin responds to Rho modification induced by bacterial toxins. Inflammasome activation leads to caspase-1 activation that in turn cleaves its downstream effectors: the newly identified pyroptosis executioner gasdermin D and pro-form of cytokines IL-1β and IL-18. DAMP, danger associated molecular pattern; GBP, guanylate binding protein; PAMP, pathogen associated molecular pattern; T3SS, type III secretion systems.”
See also Inflammasomes, the cardinal pathology mediators are activated by pathogens, allergens and mutagens: A critical review with focus on NLRP3. (2017).
What moves around within a cell upon NLRP3 inflammasome activation is very interesting: “Resting NLRP3 localizes to endoplasmic reticulum structures, whereas on inflammasome activation both NLRP3 and its adaptor ASC redistribute to the perinuclear space where they co-localize with endoplasmic reticulum and mitochondria organelle clusters.(ref).” Also, Mitochondrial dysfunction is considered crucial for NLRP3 inflammasome activation, an important point that is elaborated below.
Post-translational regulation of Inflammasomes
Because inflammasomes can create so much inflammatory havoc when activated, they are subject to multiple forms of post-translational regulation.
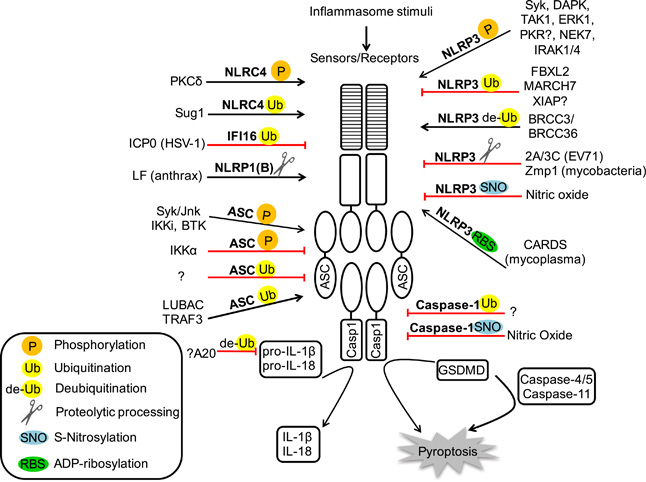
Image source: Post-translational regulation of Inflammasomes (2016)
NF-kB is important for NLRP 3 inflammasome priming
We discussed NF-kB often identified as the master regulator and initiator of inflammation, in our Part 2 blog entry for this series on inflammation. Is interesting to note that this role of NF-kB is reinforced rather than reduced when we bring inflammasomes into the inflammation picture.
Ref: Initiation and perpetuation of NLRP3 inflammasome activation and assembly. (2015) “ The NLRP3 (NOD-like receptor family, pyrin domain containing 3) inflammasome is a multiprotein complex that orchestrates innate immune responses to infection and cell stress through activation of caspase-1 and maturation of inflammatory cytokines pro-interleukin-1β (pro-IL-1β) and pro-IL-18. Activation of the inflammasome during infection can be protective, but unregulated NLRP3 inflammasome activation in response to non-pathogenic endogenous or exogenous stimuli can lead to unintended pathology. NLRP3 associates with mitochondria and mitochondrial molecules, and activation of the NLRP3 inflammasome in response to diverse stimuli requires cation flux, mitochondrial Ca(2+) uptake, and mitochondrial reactive oxygen species accumulation. It remains uncertain whether NLRP3 surveys mitochondrial integrity and senses mitochondrial damage, or whether mitochondria simply serve as a physical platform for inflammasome assembly. The structure of the active, caspase-1-processing NLRP3 inflammasome also requires further clarification, but recent studies describing the prion-like properties of ASC have advanced the understanding of how inflammasome assembly and caspase-1 activation occur while raising new questions regarding the propagation and resolution of NLRP3 inflammasome activation. Here, we review the mechanisms and pathways regulating NLRP3 inflammasome activation, discuss emerging concepts in NLRP3 complex organization, and expose the knowledge gaps hindering a comprehensive understanding of NLRP3 activation.)” The following diagram is from this publication:
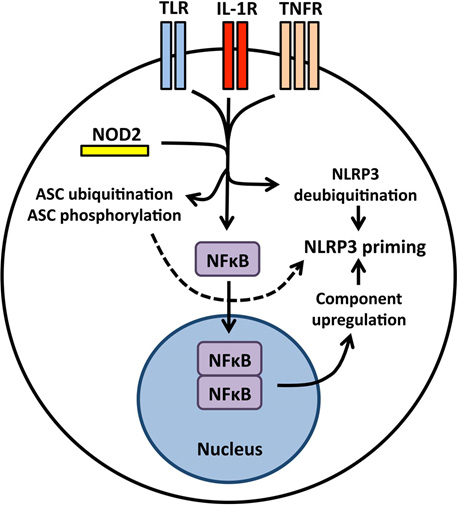
“Priming the NLRP3 inflammasome for activation NLRP3 inflammasome priming is accomplished by NFκB-activating receptors including Toll-like receptors, interleukin-1 receptor, tumor necrosis factor receptor, and the cytosolic PRR NOD2. Nuclear translocation of NFκB leads to increased synthesis of NLRP3 and inflammasome-dependent cytokine pro-IL-1β. Priming also induces post-translational modifications to inflammasome components including NLRP3 deubiqutination and ASC ubiquitination and phosphorylation.”
The very practical message is therefore: existence of inflammasomes or not, NFκB plays a critical role in the initiation of inflammation, and therefore practical control of the expression of NFκB via the dietary intake of anti-inflammatory herbs may be used to control chronic inflammation. Soon to come in this blog, a new entry On The Making Of A Dietary Supplement.
Nrf2 and inflammasomes
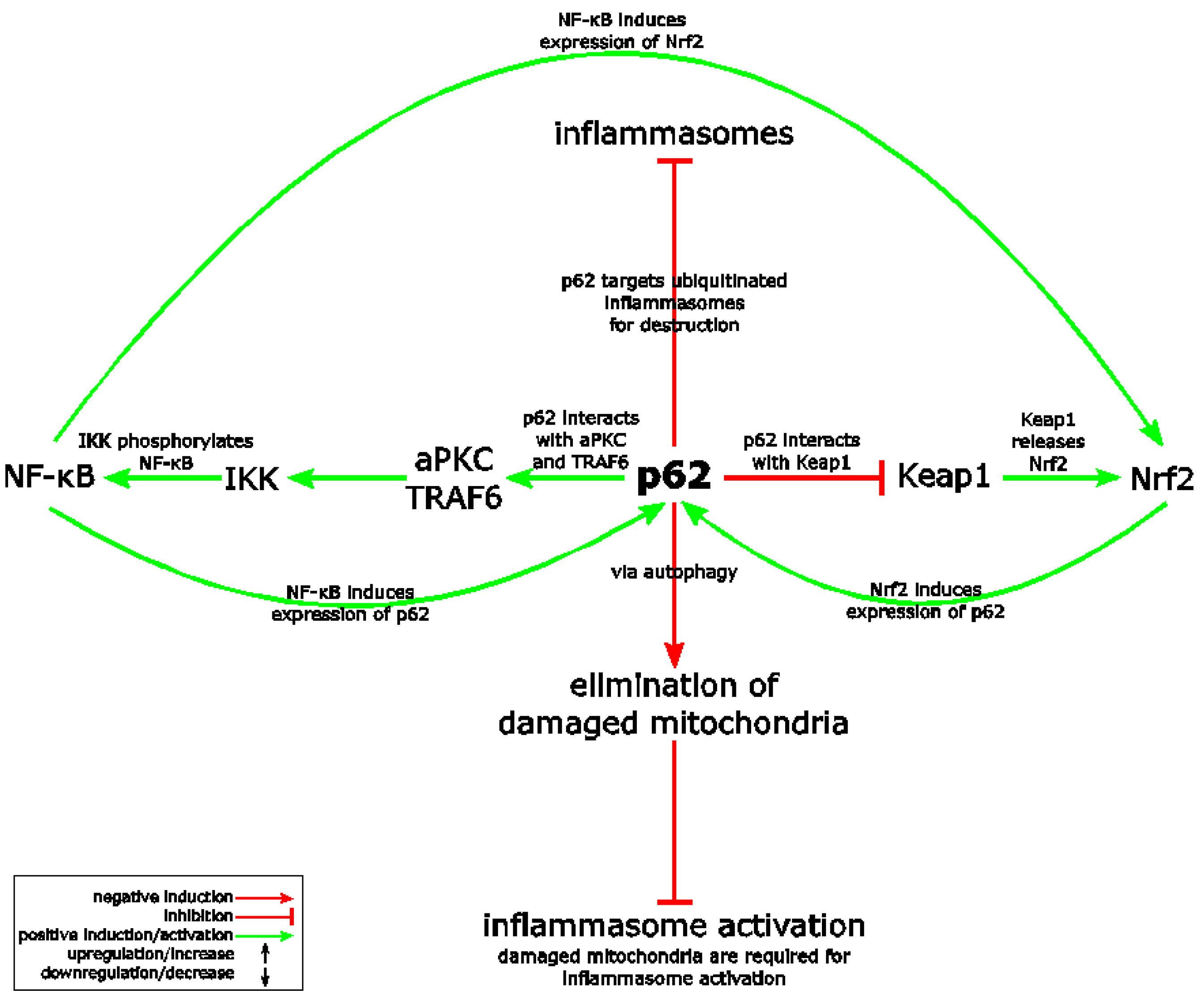
Above, we mentioned that it appears that the approaches known for limiting chronic inflammation also work to limit the formation or expression of inflammasomes. An example is the expression of Nrf2. The 2915 publication The Crosstalk between Nrf2 and Inflammasomes illustrates this observation. ““The Nrf2 (nuclear factor E2-related factor or nuclear factor (erythroid-derived 2)-like 2) transcription factor is a key player in cytoprotection and activated in stress conditions caused by reactive oxygen species (ROS) or electrophiles. Inflammasomes represent central regulators of inflammation. Upon detection of various stress factors, assembly of the inflamasome protein complex results in activation and secretion of proinflammatory cytokines. In addition, inflammasome activation causes pyroptosis, a lytic form of cell death, which supports inflammation. There is growing evidence of a crosstalk between the Nrf2 and inflammasome pathways at different levels. For example, Nrf2 activating compounds inhibit inflammasomes and consequently inflammation. This review summarizes what is known about the complex and predominantly antagonistic relationship of both stress-activated pathways.”
Here is a diagram from the publication that relates Nrf2, NF-kB, autophagy and NLRP3 inflammasome activation:
About inflammasomes and pyroptosis
By activating caspase pathways, inflammasomes can induce lyctic cell death.
Ref: The intersection of cell death and inflammasome activation. ( 2016) “Inflammasomes sense cellular danger to activate the cysteine-aspartic protease caspase-1, which processes precursor interleukin-1β (IL-1β) and IL-18 into their mature bioactive fragments. In addition, activated caspase-1 or the related inflammatory caspase, caspase-11, can cleave gasdermin D to induce a lytic cell death, termed pyroptosis. The intertwining of IL-1β activation and cell death is further highlighted by research showing that the extrinsic apoptotic caspase, caspase-8, may, like caspase-1, directly process IL-1β, activate the NLRP3 inflammasome itself, or bind to inflammasome complexes to induce apoptotic cell death. Similarly, RIPK3- and MLKL-dependent necroptotic signaling can activate the NLRP3 inflammasome to drive IL-1β inflammatory responses in vivo. Here, we review the mechanisms by which cell death signaling activates inflammasomes to initiate IL-1β-driven inflammation, and highlight the clinical relevance of these findings to heritable autoinflammatory diseases. We also discuss whether the act of cell death can be separated from IL-1β secretion and evaluate studies suggesting that several cell death regulatory proteins can directly interact with, and modulate the function of, inflammasome and IL-1β containing protein complexes.”
Additional references:
Inflammasomes as polyvalent cell death platforms. ( 2016)
Caspase-11: the driving factor for noncanonical inflammasomes. ( 2013)
A brain in flame; do inflammasomes and pyroptosis influence stroke pathology? (2017)
Pyroptotic cell death defends against intracellular pathogens (2015)
INFLAMMASOMES AND MITOCHONDRIA
We have already made a few mentions of the role mitochondria in NLRP3 inflammasome activation, such as the role of ROS produce by damaged mitochondria. However, mitochondrial misbehavior and NLRP3 inflammasome activation appeared to be tightly coupled processes. Here are a few documents that identify the mechanisms. We have italicized what we think to be particularly relevant points:
Mitochondria and the NLRP3 inflammasome: physiological and pathological relevance (2016)“ — Emerging evidence suggests that mitochondrial events are associated with NLRP3 activation in disease conditions. Mitochondrial dysfunction acts upstream of NLRP3 activation by providing reactive oxygen species (ROS) to trigger NLRP3 oligomerization or by inducing α-tubulin acetylation to relocate mitochondria to the proximity of NLRP3. In addition, mitochondria work as a platform for inflammasome assembly. Mitochondrial events may also lie downstream of NLRP3 activation. While the molecular mechanisms of mitochondrial dysfunction associated with NLRP3 activation are still unclear, they may involve the perturbation of mitochondria by K+ efflux and subsequent intracellular disequilibrium. Thus, mitochondria and NLRP3 machinery appear to be closely interwoven at multiple levels.”
Ref: A role for mitochondria in NLRP3 inflammasome activation (2011) “Previous studies suggested that NLRP3 inflammasome activity is negatively regulated by autophagy and positively regulated by reactive oxygen species (ROS) derived from an uncharacterized organelle. Here we show that mitophagy/autophagy blockade leads to the accumulation of damaged, ROS-generating mitochondria, and this in turn activates the NLRP3 inflammasome. Resting NLRP3 localizes to endoplasmic reticulum structures, whereas on inflammasome activation both NLRP3 and its adaptor ASC redistribute to the perinuclear space where they co-localize with endoplasmic reticulum and mitochondria organelle clusters. Notably, both ROS generation and inflammasome activation are suppressed when mitochondrial activity is dysregulated by inhibition of the voltage-dependent anion channel. This indicates that NLRP3 inflammasome senses mitochondrial dysfunction and may explain the frequent association of mitochondrial damage with inflammatory diseases.”
This diagram shows two dynamics that mitochondria and inflammasomes share in the development of inflammatory diseases:
![]()
Image and legend source “NLRP3 forms an inflammasome with its adaptor ASC, and its excessive activation can cause inflammatory diseases. However, little is known about the mechanisms that control assembly of the inflammasome complex. Here we show that microtubules mediated assembly of the NLRP3 inflammasome. Inducers of the NLRP3 inflammasome caused aberrant mitochondrial homeostasis to diminish the concentration of the coenzyme NAD+, which in turn inactivated the NAD+-dependent α-tubulin deacetylase sirtuin 2; this resulted in the accumulation of acetylated α-tubulin. Acetylated α-tubulin mediated the dynein-dependent transport of mitochondria and subsequent apposition of ASC on mitochondria to NLRP3 on the endoplasmic reticulum. Therefore, in addition to direct activation of NLRP3, the creation of optimal sites for signal transduction by microtubules is required for activation of the entire NLRP3 inflammasome.”
The pathway identified as Signal 1 shows mitochondrial damage resulting in reduction of NAD+ and SIRT2, accumulation of acetylated alpha-tublin, and involvement of microtubules and mitochondrial transport — topics covered in depth in other blog entries. The possibility for therapeutic interventions seems very rich, including upgrading the expression of NAD+ and steps to minimize mitochondrial damage. What a incredibly rich web of interacting pathways! I have a feeling the collectively we are beginning to get on top of them though it may take a long time for this to be complete. (Vince comment).,
We thought we understood inflammation pretty well before we knew about inflammasomes, but this last comment is indicative of the additional insight into inflammation that inflammasomes give us. A simple practical consequence may be that an excellent way to prevent spurious assembly or activation of NLRP3 inflammasomes may be to keep mitochondria healthy. Actually, it’s a bit more complex than that and circular. As pointed out in the next article quoted, inflammasomes in turn can result in damaged mitochondria.
Ref: Caspase-1–dependent mitochondrial damage and block of mitophagy (2014) “Inflammasomes are intracellular sensors that couple detection of pathogens and cellular stress to activation of Caspase-1, and consequent IL-1β and IL-18 maturation and pyroptotic cell death. Here, we show that the absent in melanoma 2 (AIM2) and nucleotide-binding oligomerization domain-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasomes trigger Caspase-1–dependent mitochondrial damage. Caspase-1 activates multiple pathways to precipitate mitochondrial disassembly, resulting in mitochondrial reactive oxygen species (ROS) production, dissipation of mitochondrial membrane potential, mitochondrial permeabilization, and fragmentation of the mitochondrial network. Moreover, Caspase-1 inhibits mitophagy to amplify mitochondrial damage, mediated in part by cleavage of the key mitophagy regulator Parkin. In the absence of Parkin activity, increased mitochondrial damage augments pyroptosis, as indicated by enhanced plasma membrane permeabilization and release of danger-associated molecular patterns (DAMPs). Therefore, like other initiator caspases, Caspase-1 activation by inflammasomes results in mitochondrial damage.” There we have it: mitochondrial damage and NLRP3 inflammasome activation feed upon each other.
Ref. Mitochondria: diversity in the regulation of the NLRP3 inflammasome (2015) Ref. Bottom-line summary: 1 “Mitochondrial dysfunction induces specific activation of the NLRP3 inflammasome. 2. Mitochondria recruit NLRP3 and aid the assembly of the NLRP3 inflammasome. 3. Autophagy/mitophagy clears damaged mitochondria and induces inflammasome activation. 4. Mitochondria and NLRP3 dysfunction promotes metabolic diseases.”
Ref. Role of mitochondrial dysfunction in NLRP3 inflammasome (2015) “Mitochondrial dysfunction is considered crucial for NLRP3 inflammasome activation partly through its release of mitochondrial toxic products such as mitochondrial ROS (mROS) and mitochondrial DNA (mtDNA). While previous studies have shown that classical NLRP3-activating stimulations lead to mROS generation and mtDNA release, it remains poorly understood whether and how mitochondrial damage-derived factors may contribute to NLRP3 inflammasome activation. Here, we demonstrate that impairment of the mitochondrial electron transport chain by rotenone licenses NLRP3 inflammasome activation only upon costimulation with ATP, but not with nigericin or alum. Rotenone-induced priming of NLRP3 in the presence of ATP triggered the formation of speck-like NLRP3 or ASC aggregates and the association of NLRP3 with ASC, resulting in NLRP3-dependent caspase-1 activation. Mechanistically, rotenone confers a priming signal for NLRP3 inflammasome activation only in the context of aberrant high-grade, but not low-grade, mROS production and mitochondrial hyperpolarization. By contrast, rotenone/ATP-mediated mtDNA release and mitochondrial depolarization are likely to be merely an indication of mitochondrial damage rather than triggering factors for NLRP3 inflammasome activation. Our results provide a molecular insight into the selective contribution made by mitochondrial dysfunction to the NLRP3 inflammasome pathway.”
Activation of the NLRP3 inflammasome can lead to mitochondrial damage
Ref. Inflammasome activation leads to Caspase-1–dependent mitochondrial damage and block of mitophagy (2014) “Sensors of the innate immune system trigger the induction of inflammatory responses upon infection and cellular stress. One such sensor is the inflammasome pathway, which is best described for its role in the production of the inflammatory cytokines IL-1β and IL-18. Other effector functions of the inflammasome pathway remain less well defined, and here we show that activation of this pathway leads to induction of mitochondrial damage and dismantling of the organelle. We link such mitochondrial damage to another inflammasome-regulated process called pyroptosis, which is a proinflammatory form of cell death. In summary, we characterize the role of mitochondrial damage during activation of the inflammasome pathway, of relevance to host defense and other physiological and pathophysiological settings. – Inflammasomes are intracellular sensors that couple detection of pathogens and cellular stress to activation of Caspase-1, and consequent IL-1β and IL-18 maturation and pyroptotic cell death. Here, we show that the absent in melanoma 2 (AIM2) and nucleotide-binding oligomerization domain-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasomes trigger Caspase-1–dependent mitochondrial damage. Caspase-1 activates multiple pathways to precipitate mitochondrial disassembly, resulting in mitochondrial reactive oxygen species (ROS) production, dissipation of mitochondrial membrane potential, mitochondrial permeabilization, and fragmentation of the mitochondrial network. Moreover, Caspase-1 inhibits mitophagy to amplify mitochondrial damage, mediated in part by cleavage of the key mitophagy regulator Parkin. In the absence of Parkin activity, increased mitochondrial damage augments pyroptosis, as indicated by enhanced plasma membrane permeabilization and release of danger-associated molecular patterns (DAMPs). Therefore, like other initiator caspases, Caspase-1 activation by inflammasomes results in mitochondrial damage.”
Ref. Mitochondrial function is required for extracellular ATP-induced NLRP3 inflammasome activation 2017 “Accumulating evidence suggests that mitochondria are common mediators of NLRP3 inflammasomeactivation induced by a wide range of inflammatory stimuli — “.
Mitochondrial dysfunction, inflammasomes, chronic inflammation and aging
The processes outlined just above are extremely relevant to a central topic in this blog series, chronic inflammation. The 2012 publication Mitochondrial dysfunction and oxidative stress activate inflammasomes: Impact on the aging process and age-related diseases summarizes the situation: “Oxidative stress and low-grade inflammation are the hallmarks of the aging process and are even more enhanced in many age-related degenerative diseases. Mitochondrial dysfunction and oxidative stress can provoke and potentiate inflammatory responses, but the mechanism has remained elusive. Recent studies indicate that oxidative stress can induce the assembly of multiprotein inflammatory complexes called the inflammasomes. Nod-like receptor protein 3 (NLRP3) is the major immune sensor for cellular stress signals, e.g., reactive oxygen species, ceramides, and cathepsin B. NLRP3 activation triggers the caspase-1-mediated maturation of the precursors of IL-1β and IL-18 cytokines. During aging, the autophagic clearance of mitochondria declines and dysfunctional mitochondria provoke chronic oxidative stress, which disturbs the cellular redox balance. Moreover, increased NF-κB signaling observed during aging could potentiate the expression of NLRP3 and cytokine proforms enhancing the priming of NLRP3 inflammasomes. Recent studies have demonstrated that NLRP3 activation is associated with several age-related diseases, e.g., the metabolic syndrome. We will review here the emerging field of inflammasomes in the appearance of the proinflammatory phenotype during the aging process and in age-related diseases.”A number of additional publications dealing with these points including the following references:
Mitochondria play a central role in NLRP3 inflammasome activation (2014)
Mitochondria: diversity in the regulation of the NLRP3 inflammasome (2015)
Inflammasome Activation by Mitochondrial Oxidative Stress in Macrophages Leads to the Development of Angiotensin. II–Induced Aortic Aneurysm (2015)
Mitophagy: a balance regulator of NLRP3 inflammasome activation (2016)
Mitochondrial control of the NLRP3 inflammasome
INFLASOMES AND DISEASE PROCESSESA great many publications have focused on the roles of inflammasome’s in various disease processes. Typical examples of these follow.1. Inflammasome and AtherosclerosisThe NLRP3 Inflammasome subtype links cholesterol crystals to the formation of atherosclerotic “vasculitis.”Ref. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals (2010) “The inflammatory nature of atherosclerosis is well established but the agent(s) that incite inflammation in the artery wall remain largely unknown. Germ-free animals are susceptible to atherosclerosis, suggesting that endogenous substances initiate the inflammation1. Mature atherosclerotic lesions contain macroscopic deposits of cholesterol crystals in the necrotic core, but their appearance late in atherogenesis had been thought to disqualify them as primary inflammatory stimuli. However, using a new microscopic technique, we revealed that minute cholesterol crystals are present in early diet-induced atherosclerotic lesions and that their appearance in mice coincides with the first appearance of inflammatory cells. Other crystalline substances can induce inflammation by stimulating the caspase-1-activating NLRP3 (NALP3 or cryopyrin) inflammasome2,3, which results in cleavage and secretion of interleukin (IL)-1 family cytokines. Here we show that cholesterol crystals activate the NLRP3 inflammasome in phagocytes in vitro in a process that involves phagolysosomal damage. Similarly, when injected intraperitoneally, cholesterol crystals induce acute inflammation, which is impaired in mice deficient in components of the NLRP3 inflammasome, cathepsin B, cathepsin L or IL-1 molecules. Moreover, when mice deficient in low-density lipoprotein receptor (LDLR) were bone-marrow transplanted with NLRP3-deficient, ASC (also known as PYCARD)-deficient or IL-1α/β-deficient bone marrow and fed on a high-cholesterol diet, they had markedly decreased early atherosclerosis and inflammasome-dependent IL-18 levels. Minimally modified LDL can lead to cholesterol crystallization concomitant with NLRP3 inflammasome priming and activation in macrophages. Although there is the possibility that oxidized LDL activates the NLRP3 inflammasome in vivo, our results demonstrate that crystalline cholesterol acts as an endogenous danger signal and its deposition in arteries or elsewhere is an early cause rather than a late consequence of inflammation. These findings provide new insights into the pathogenesis of atherosclerosis and indicate new potential molecular targets for the therapy of this disease.”2. Inflammasomes, Insulin Resistance, and Type II Diabetes
The NLRP3 Inflammasome links obesity, free fatty acids, and ceremides to obesity-induced inflammation and insulin resistance. This explains many features of metabolic syndrome and T2DM not well explained by protein glycation.
Ref. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance (2011) “The emergence of chronic inflammation during obesity in the absence of overt infection or well-defined autoimmune processes is a puzzling phenomenon. The Nod-like receptor (NLR) family of innate immune cell sensors, such as the nucleotide-binding domain, leucine-rich–containing family, pyrin domain–containing-3 (Nlrp3, but also known as Nalp3 or cryopyrin) inflammasome are implicated in recognizing certain nonmicrobial originated ‘danger signals’ leading to caspase-1 activation and subsequent interleukin-1β (IL-1β) and IL-18 secretion. We show that calorie restriction and exercise-mediated weight loss in obese individuals with type 2 diabetes is associated with a reduction in adipose tissue expression of Nlrp3 as well as with decreased inflammation and improved insulin sensitivity. We further found that the Nlrp3 inflammasome senses lipotoxicity-associated increases in intracellular ceramide to induce caspase-1 cleavage in macrophages and adipose tissue. Ablation of Nlrp3 in mice prevents obesity-induced inflammasome activation in fat depots and liver as well as enhances insulin signaling. Furthermore, elimination of Nlrp3 in obese mice reduces IL-18 and adipose tissue interferon-γ (IFN-γ) expression, increases naive T cell numbers and reduces effector T cell numbers in adipose tissue. Collectively, these data establish that the Nlrp3 inflammasome senses obesity-associated danger signals and contributes to obesity-induced inflammation and insulin resistance.”
3. The NLRP3 inflammasome and neuroinflammation
it appears that Inflammasomes play important roles in practically every disease condition the involves neuroinflammation, including autoimmune diseases. A small sampling from the literature follows:
Ref. NLRP3 Inflammasome in Neurological Diseases, from Functions to Therapies (2017)
“Neuroinflammation has been identified as a causative factor of multiple neurological diseases. The nucleotide-binding oligomerization domain-, leucine-rich repeat- and pyrin domain-containing 3 (NLRP3) inflammasome, a subcellular multiprotein complex that is abundantly expressed in the central nervous system (CNS), can sense and be activated by a wide range of exogenous and endogenous stimuli such as microbes, aggregated and misfolded proteins, and adenosine triphosphate, which results in activation of caspase-1. Activated caspase-1 subsequently leads to the processing of interleukin-1β (IL-1β) and interleukin-18 (IL-18) pro-inflammatory cytokines and mediates rapid cell death. IL-1β and IL-18 drive inflammatory responses through diverse downstream signaling pathways, leading to neuronal damage. Thus, the NLRP3 inflammasome is considered a key contributor to the development of neuroinflammation. In this review article, we briefly discuss the structure and activation the NLRP3 inflammasome and address the involvement of the NLRP3 inflammasome in several neurological disorders, such as brain infection, acute brain injury and neurodegenerative diseases. In addition, we review a series of promising therapeutic approaches that target the NLRP3 inflammasome signaling including anti-IL-1 therapy, small molecule NLRP3 inhibitors and other compounds, however, these approaches are still experimental in neurological diseases. At present, it is plausible to generate cell-specific conditional NLRP3 knockout (KO) mice via the Cre system to investigate the role of the NLRP3 inflammasome, which may be instrumental in the development of novel pharmacologic investigations for neuroinflammation-associated diseases.”
See also references:
4. The NLRP3 inflammasome and age-related macular degeneration (AMD)
There is a large number of published articles on this topic. Activation of the NLRP3 inflammasome, along with complement activation and oxidative damage, have been implicated in retinal pigment epithelium (RPE) pathology in age-related macular degeneration (AMD) Further, there is speculation that AMD may be controlled by plant-based dietary substances that can regulate inflammasome formation and activation. You can start with this review article:
NLRP3 inflammasome: activation and regulation in age-related macular degeneration. (2015)
“Age-related macular degeneration (AMD) is the leading cause of legal blindness in the elderly in industrialized countries. AMD is a multifactorial disease influenced by both genetic and environmental risk factors. Progression of AMD is characterized by an increase in the number and size of drusen, extracellular Ref: deposits, which accumulate between the retinal pigment epithelium (RPE) and Bruch’s membrane (BM) in outer retina. The major pathways associated with its pathogenesis include oxidative stress and inflammation in the early stages of AMD. Little is known About the interactions among these mechanisms that drive the transition from early to late stages of AMD, such as geographic atrophy (GA) or choroidal neovascularization (CNV). As part of the innate immune system, inflammasome activation has been identified in RPE cells and proposed to be a causal factor for RPE dysfunction and degeneration. Here, we will first review the classic model of inflammasome activation, then discuss the potentials of AMD-related factors to activate the inflammasome in both nonocular immune cells and RPE cells, and finally introduce several novel mechanisms for regulating the inflammasome activity.”
See also references:
VEGF-A and the NLRP3 Inflammasome in Age-Related Macular Degeneration Marneros AG, et al. Adv Exp Med Biol. 2016
NLRP3 inflammasome blockade inhibits VEGF-A-induced age-related macular degeneration. Marneros AG, et al. Cell Rep. 2013
NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: implications for age-related macular degeneration.Tseng WA, et al. Invest Ophthalmol Vis Sci. 2013
NLRP3 inflammasome: activation and regulation in age-related macular degeneration. Gao J, et al. Mediators Inflamm. 2015
Complement Component C5a Primes Retinal Pigment Epithelial Cells for Inflammasome Activation by Lipofuscin-mediated Photooxidative Damage. Brandstetter C, et al. J Biol Chem. 2015
NLRP3 Upregulation in Retinal Pigment Epithelium in Age-Related Macular Degeneration. Wang Y, et al. Int J Mol Sci. 2016
A2E induces IL-1ß production in retinal pigment epithelial cells via the NLRP3 inflammasome. Anderson OA, et al. PLoS One. 2013
Light induces NLRP3 inflammasome activation in retinal pigment epithelial cells via lipofuscin-mediated photooxidative damage. Brandstetter C, et al. J Mol Med (Berl). 2015
Increased VEGF-A promotes multiple distinct aging diseases of the eye through shared pathomechanisms. Marneros AG, et al. EMBO Mol Med. 2016
Amyloid β induces NLRP3 inflammasome activation in retinal pigment epithelial cells via NADPH oxidase- and mitochondria-dependent ROS production. Wang K, et al. J Biochem Mol Toxicol. 2017
Key messages appear to be: “• Visible light irradiation of lipofuscin-loaded RPE cells activates inflammasome. • Inflammasome activation results from lysosomal permeabilization and enzyme leakage. • Inflammasome activation induces secretion of inflammatory cytokines by RPE cells. • Photooxidative damage by visible light as new mechanism of inflammasome activation. • Novel link between hallmark pathogenetic features of retinal degenerative diseases.(ref).”
5. Central nervous system bacterial infection
We can easily fall into thinking of inflammasomes as evil assemblages which lock in chronic inflammation. This can lead us to forget their evolutionary role of protecting us against numerous potentially deadly assaults. One such assault, for example, can be Staphylococcus aureus (S. aureus) infection in the central nervous system (CNS), which can be fatal.
Ref. Critical role for the AIM2 inflammasome during acute CNS bacterial infection. (2014) “Interleukin-1β (IL-1β) is essential for eliciting protective immunity during the acute phase of Staphylococcus aureus (S. aureus) infection in the central nervous system (CNS). We previously demonstrated that microglial IL-1β production in response to live S. aureus is mediated through the Nod-like receptor protein 3 (NLRP3) inflammasome, including the adapter protein ASC (apoptosis-associated speck-like protein containing a caspase-1 recruitment domain), and pro-caspase 1. Here, we utilized NLRP3, ASC, and caspase 1/11 knockout (KO) mice to demonstrate the functional significance of inflammasome activity during CNS S. aureus infection. ASC and caspase 1/11 KO animals were exquisitely sensitive, with approximately 50% of mice succumbing to infection within 24 h. Unexpectedly, the survival of NLRP3 KO mice was similar to wild-type animals, suggesting the involvement of an alternative upstream sensor, which was later identified as absent in melanoma 2 (AIM2) based on the similar disease patterns between AIM2 and ASC KO mice. Besides IL-1β, other key inflammatory mediators, including IL-6, CXCL1, CXCL10, and CCL2 were significantly reduced in the CNS of AIM2 and ASC KO mice, implicating autocrine/paracrine actions of IL-1β, as these mediators do not require inflammasome processing for secretion. These studies demonstrate a novel role for the AIM2 inflammasome as a critical molecular platform for regulating IL-1β release and survival during acute CNS S. aureus infection.
A similar protective pathway is activated by the Myxoma virus:
Ref. Myxoma virus lacking the pyrin-like protein M013 is sensed in human myeloid cells by both NLRP3 and multiple Toll-like receptors, which independently activate the inflammasome and NF-κB innate response pathways. ( 2011) “The myxoma virus (MYXV)-encoded pyrin domain-containing protein M013 coregulates inflammatory responses mediated by both the inflammasome and the NF-κB pathways. Infection of human THP-1 monocytic cells with a MYXV construct deleted for the M013 gene (vMyxM013-KO), but not the parental MYXV, activates both the inflammasome and NF-κB pathways and induces a spectrum of proinflammatory cytokines and chemokines, like interleukin-1β (IL-1β), tumor necrosis factor (TNF), IL-6, and monocyte chemoattractant protein 1. Here, we report that vMyxM013-KO virus-mediated activation of inflammasomes and secretion of IL-1β are dependent on the adaptor protein ASC, caspase-1, and NLRP3 receptor. However, vMyxM013-KO virus-mediated activation of NF-κB signaling, which induces TNF secretion, was independent of ASC, caspase-1, and either the NLRP3 or AIM2 inflammasome receptors. We also report that early synthesis of pro-IL-1β in response to vMyxM013-KO infection is dependent upon the components of the inflammasome complex. Activation of the NLRP3 inflammasome and secretion of IL-1β was also dependent on the release of cathepsin B and production of reactive oxygen species (ROS). By using small interfering RNA screening, we further demonstrated that, among the RIG-I-like receptors (RLRs) and Toll-like receptors (TLRs), only TLR2, TLR6, TLR7, and TLR9 contribute to the NF-κB-dependent secretion of TNF and the inflammasome-dependent secretion of IL-1β in response to vMyxM013-KO virus infection. Additionally, we demonstrate that early triggering of the mitogen-activated protein kinase pathway by vMyxM013-KO virus infection of THP-1 cells plays a critical common upstream role in the coordinate induction of both NF-κB and inflammasome pathways. We conclude that an additional cellular sensor(s)/receptor(s) in addition to the known RLRs/TLRs plays a role in the M013 knockout virus-induced activation of NF-κB pathway signaling, but the activation of inflammasomes entirely depends on sensing by the NLRP3 receptor in response to vMyxM013-KO infection of human myeloid cells.”
See also references:
The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. (2010) “These results suggest that NLRP3 plays an important role in a model of multiple sclerosis by exacerbating CNS inflammation, and this is partly mediated by caspase-1 and IL-18”
Inflammasome activation in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). (2017) “Experiments with the mouse model of MS, experimental autoimmune encephalomyelitis (EAE), specifically describe the NLRP3 inflammasome as critical and necessary to disease development.”
Ref. Critical role for The AIM2 inflammasome during acute CNS bacterial infection. (2014)
6. Inflammasome’s in Alzheimer’s disease
They play important roles and may offer therapeutic pathways.
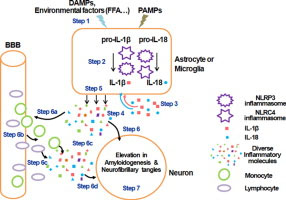
Ref. The role of inflammasome in Alzheimer’s disease (2016)
“Highlights: Inflammasomes involved in IL-1β and IL-18 maturation contribute to AD progression, Environmental factors, i.e. fatty acids, can trigger the activation of inflammasome., Modulating inflammasome activation could be a potential strategy for treating AD. Alzheimer’s disease (AD) is a chronic, progressive and irreversible neurodegenerative disease with clinical characteristics of memory loss, dementia and cognitive impairment. Although the pathophysiologic mechanism is not fully understood, inflammation has been shown to play a critical role in the pathogenesis of AD. Inflammation in the central nervous system (CNS) is characterized by the activation of glial cells and release of proinflammatory cytokines and chemokines. Accumulating evidence demonstrates that inflammasomes, which cleave precursors of interleukin-1β (IL-1β) and IL-18 to generate their active forms, play an important role in the inflammatory response in the CNS and in AD pathogenesis. Therefore, modulating inflammasome complex assembly and activation could be a potential strategy for suppressing inflammation in the CNS. This review aims to provide insight into the role of inflammasomes in the CNS, with respect to the pathogenesis of AD, and may provide possible clues for devising novel therapeutic strategies.”
Graphical abstract for the above:
See also references:
Inflammasome activation and innate immunity in Alzheimer’s disease. (2017) .
Inflammasomes as therapeutic targets for Alzheimer’s disease. (2017)
The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. (2016)
Amyloid-beta oligomers set fire to inflammasomes and induce Alzheimer’s pathology. ( 2008)
7. Inflammasomes and multiple sclerosis
Ref: Inflammasome activation in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). (2017) “The aptly named inflammasomes are powerful signaling complexes that sense inflammatory signals under a myriad of conditions, including those from infections and endogenous sources. The inflammasomes promote inflammation by maturation and release of the pro-inflammatory cytokines, IL-1β and IL-18. Several inflammasomes have been identified so far, but this review focuses mainly on the NLRP3 inflammasome. By still ill-defined activation mechanisms, a sensor molecule, NLRP3 (NACHT, LRR and PYD domains-containing protein 3), responds to danger signals and rapidly recruits ASC (apoptosis-associated speck-like protein containing a CARD) and pro-caspase-1 to form a large oligomeric signaling platform-the inflammasome. Involvement of the NLRP3 inflammasome in infections, metabolic disorders, autoinflammation, and autoimmunity, underscores its position as a central player in sensing microbial and damage signals and coordinating pro-inflammatory immune responses. Indeed, evidence in patients with multiple sclerosis (MS) suggests inflammasome activation occurs during disease. Experiments with the mouse model of MS, experimental autoimmune encephalomyelitis (EAE), specifically describe the NLRP3 inflammasome as critical and necessary to disease development. This review discusses recent studies in EAE and MS which describe associations of inflammasome activation with promotion of T cell pathogenicity, infiltration of cells into the central nervous system (CNS) and direct neurodegeneration during EAE and MS.”
Ref: The role of interferon-β in the treatment of multiple sclerosis and experimental autoimmune encephalomyelitis – in the perspective of inflammasomes. (2013) Review.
8. Additional publications relating to mechanisms of inflamasomes and of The relationship of inflammasomes to disease processes
We mostly exclude publications that are primarily focused on negative regulation of inflammasomes here, but plan to list many of them in the Part 6 blog entry.
Research progress on the NLRP3 inflammasome and its role in the central nervous system. 2013
[Inflammatory bowel diseases and inflammasome]. 2011
The NLRP3 inflammasome in kidney disease and autoimmunity. 2016
NLRP3 inflammasome and host protection against bacterial infection. 2013
Interferons and inflammasomes: Cooperation and counterregulation in disease. 2016
.Negative regulation of NLRP3 inflammasome signaling.
The inflammasome: an emerging therapeutic oncotarget for cancer prevention. 2016
Negative regulators and their mechanisms in NLRP3 inflammasome activation and signaling. 2017
The inflammasome and lupus: another innate immune mechanism contributing to disease pathogenesis?. 2014
Activation and regulation of the inflammasomes. 2013 Review.
NLRP3 inflammasome-driven pathways in depression: Clinical and preclinical findings. 2017
The nucleic acid-sensing inflammasomes..
P2X7R/cryopyrin inflammasome axis inhibition reduces neuroinflammation after SAH. 2013
NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes.. 2015
Initiation and perpetuation of NLRP3 inflammasome activation and assembly. 2015
P2X7R/cryopyrin inflammasome axis inhibition reduces neuroinflammation after SAH.. 2013
——————————————————————————
Again, we expect that Part 6 of this series on inflammation will be on Substances That Can Limit Or Prevent The Formation Or Expression Of Inflammasomes.
—————————————————————————————
Show details |

Pingback: Inflammation Part 4 - PCSK9 inhibition - Also Part 1 of interventions that reduce all cause mortality (ACM) - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™
Pingback: Inflammation Part 8: Neurohormesis, neuroinflammatory diseases, and their treatment by mushroom substances (Section 2 of this discussion) Can a mushroom play an important role in the health and anti-aging picture? Recent research says Yes, and this blog e
Pingback: Inflammation Part 7: Neurohormesis, neuroinflammatory diseases, and their treatment by mushroom substances (Section 1) - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™