
By Vince Giuliano
INTRODUCTION
Many of us who have studied aging have long believed that, sooner or later we would discover keys to a long and healthy life via biological repair, renewal and regenerative processes. Based on what we have been learning very recently, the real possibility is sooner. These keys include DNA repair, effective mobilization of stem cells, and the possibility of restoring cells to earlier epigenetic states. We know now that seriously damaged organs can be renovated. Further, deep understanding of these processes and effective interventions related to them may allow us to significantly extend our lifespans and health spans. We have also been deeply concerned with understanding certain processes of aging which we have regarded to be deleterious and worth fighting, including inflammation and the SASP, what happens when old senescent cells do not go away but instead become bad citizens. Now we know they are not all bad, and indeed may be harnessed in the interest of health and longevity.’
Further, deep understanding of these processes and effective interventions related to them may allow us to significantly extend our lifespans and health spans. We have also been deeply concerned with understanding certain processes of aging which we have regarded to be deleterious and worth fighting, including inflammation and the SASP, what happens when old senescent cells do not go away but instead become bad citizens. Now we know they are not all bad, and indeed may be harnessed in the interest of health and longevity.’
We are entering an era where organ repair, renewal and regeneration has been demonstrated in laboratory fish and small animals, where we are understanding the molecular mechanisms through which these can take place, and we know how to trigger them. We know a substance that can repair and renew seriously damaged hearts in zebrafish and mice, and grow new fins on zebrafish. We know how a serious injury can trigger de-differentiation of neighboring cells to earlier epigenetic stem-cell like states, where these cells can proceed to regenerate complex organs. This blog entry reports on these exciting developments.
The basic message in a nutshell
When we are young and peppy, cell and organ regeneration and renewal go on as background processes in our body all the time. Here is how it works: molecular distress signals such as associated with injuries, inflammation, or cell senescence, trigger the partial regression of normal mature body cells back to earlier less differentiated epigenetic states. This is a process of de-differentiation, one that moves cells backwards in their developmental trajectory towards becoming stem cell, but that stops short of going all the way. Then these de-differentiated cells re-differentiate so as to create healthy new cells. And renew organs, as needed. If the injury is to cells that are supposedly permanent and do not reproduce, such as neurons in the brain, no problem. In this case neighboring microglial de-differentiate and then re-differentiate changing their identity to becoming neurons. The de-differentiation takes place with the body using the same factors that Yamanaka and other researchers have used to de-differentiate cells in-vitro removed from the body: Oct3/4, Sox2, Klf4, and c-Myc, commonly referred to as OSKM. All this happens on the local level when needed, whether within the heart, the brain, the liver or elsewhere. As we grow older, however, typical changes occur that effectively slows down or stops this renewal process. These include certain epigenetic changes associated with aging, and states of chronic inflammation. These inhibiting factors appear to be only partially understood as of now. Despite this, the very good news appears to be that to a large extent we may be able to control and reverse the adverse epigenetic changes and neutralize chronic inflammation. We already know the interventions that do this, and they are very simple and broadly available. We don’t need additional scientific breakthroughs; we don’t need extremely expensive new drugs or high-technology treatments. This may be the blockbuster news with regard to antiaging approaches that we have been waiting for for so many years. Here, now.
FIRST, ABOUT AGING
A good argument can be made that aging is the result of decline and loss of tissue homeostasis, that is loss of capability tissues and organs to maintain and repair themselves. An excellent article published in December 2016 is In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. “Embryonic development occurs as a unidirectional progression from a single-cell zygote to an adult organism. During embryogenesis and early stages of life, cells undergo a spatiotemporally orchestrated differentiation process, leading to the generation of all of the cell types that comprise an adult organism. These events take place within a stable environment that minimizes molecular and cellular damage. As an organism ages, however, there is a continuous and progressive decline in the mechanisms responsible for minimizing cellular damage.” In this and many other blog entries, I suggest that there may be some simple and powerful things we can do prevent and possibly reverse that decline.
Of high relevance to the current blog entry is the following paragraph relating background from the same publication: “The last decade of scientific research has dramatically improved our understanding of the aging process (Johnson et al., 2013; Kenyon, 2010; Riera et al., 2016). The notion that cells undergo a unidirectional differentiation process during development was proved wrong by the experimental demonstration that a terminally differentiated cell can be reprogrammed into a pluripotent embryonic-like state (Gurdon, 1962; Takahashi and Yamanaka, 2006). Cellular reprogramming to pluripotency by forced expression of the Yamanaka factors (Oct4, Sox2, Klf4, and c-Myc [OSKM]) occurs through the global remodeling of epigenetic marks (Buganim et al., 2012, 2013; Hansson et al., 2012; Polo et al., 2012). Importantly, many of the epigenetic marks that are remodeled during reprogramming (e.g., DNA methylation, post-translational modification of histones, and chromatin remodeling) are dysregulated during aging (Benayoun et al., 2015; Liu et al., 2013b; Pollina and Brunet, 2011). In fact, epigenetic dysregulation has emerged as a key hallmark of the aging process (Sen et al., 2016). Several groups, including ours, have observed an amelioration of age-associated cellular phenotypes during in vitro cellular reprogramming (Lapasset et al., 2011; Liu et al., 2011; Mahmoudi and Brunet, 2012; Rando and Chang, 2012). Reprogramming of cells from centenarians or patients with Hutchinson-Gilford progeria syndrome, (HGPS) a disorder characterized by premature aging, resets telomere size, gene expression profiles, and levels of oxidative stress, resulting in the generation of rejuvenated cells (Lapasset et al., 2011; Liu et al., 2011; Zhang et al., 2011). — Together, these observations suggest that cellular reprogramming may be used to promote tissue regeneration and led us to hypothesize that in vivo partial reprogramming could slow or reverse the aging process and extend organismal lifespan.”
In this blog entry I cite recent research publications which collectively suggest that our bodies lose a natural endogenous capability for tissue and organ repair due to systemic inflammation and certain dysregulated epigenetic factors, and this may be responsible for most diseases of old age. If this perspective is correct, it could constitute incredibly good news for old people like me. And that is because we are beginning to understand things we can do to prevent systematic inflammation and alter some of those dysregulated epigenetic factors. Follow this story.
Bad biological processes that happen with aging are sometimes good and essential
Research in just the last year or two has been yielding a much deeper understanding of the relationships between the old-age phenomena of chronic inflammation, the SASP (senescence-associated secretory phenotype) and repair and regeneration on the molecular level. This is causing us to reject or revise many of the simplistic concepts we have held in favor of much more complex nuanced ones. As a teaser specifically, the presence of both inflammation and the SASP is required to trigger repair and rejuvenation processes. These are good things to have a little of under the right circumstances, bad things to have too much of or under the wrong circumstances. And we were traditionally wrong about many other things as well, such as the dogma that neurons never regenerate and that if you lose some of the course of life, well that is just too bad for you.
In this blog I will discuss findings of several recent publications linking up the four topics (biological repair and regeneration, inflammation, the SASP and aging), where the related research appears to be going, and very simple practical implications for old folks like me. These discoveries relate to circle of life phenomena, that is, molecular phenomena that are absolutely critical in embryogenesis and very early development of life, that have largely been evolutionarily conserved over a broad variety of organisms, that have traditionally been viewed as detrimental in old age (the theory of antagonistic pleiotropy), but that can probably be much more effectively harnessed to provide renewal and revitalization at any age.
A great many past blog entries are antecedents to and provide background to this one. There is our recent ongoing blog series on inflammation for example.
Biological processes tend to be organized according to overlapping networks rather than according to hierarchical structures. As we have often preached, there is no center to biology or biological processes. As usual, the literature concerned tends to consist of articles dealing only with specific factors, usually ignoring many others that are relevant. So, how best to organize this discussion? I will start with short recaps of individual topics of central concern.
REPAIR AND REJUVENATION
Here, we have a variety of phenomena that have been studied extensively, examples of which include wound healing, stem cell activity, and DNA repair. A broad generalization is that the same or similar processes are involved in the early stages of life, and in life going on at all ages. However, repair and rejuvenation processes tend to become less effective with advanced age.
A naïve approach to organ regeneration which did not work
Let’s start with in-situ regeneration. A number of years ago, I first wrote in this blog about induced pluripotent stem cells (ref)(ref)(ref). I suggested a possible approach to a very long life, which I referred to as closing the loop in the stem cell supply chain (ref)(ref)(ref). This approach would involve, first, removal of normal somatic cells from the body, second reverting them to being pluripotent stem cells using some variation of the Yamanaka renewal factors, and third, reintroduction of the pluripotent stem cells into appropriate niches in the body. There, they would be able to re-differentiate into tissues that repair and replace damaged ones The Yamanaka factors were known to be effective in inducing somatic cell to regress to essentially having original pluripotent stem cell properties. The original factors were of course Oct3/4, Sox2, Klf4, and c-Myc, commonly referred to as OSKM. This approach did not pan out, the basic problem being in the final step, effective reintroducing completely pluripotent stem cells into their original niches. In experiments, cells were completely regressed to near original pluripotent stem cell status, and had been wiped out of any memory of their original cell lineage. Often when introduced into the body, they would induce teratomas, that is cysts consisting of mixed tissue which had nothing to do with each other or with what the tissues are supposed to be the point of insertion, where introduced into the body. Teratomas might, for example, contain teeth, nerve, heart, hair and muscle fibers.
Reference: Reprogramming in vivo produces teratomas and iPS cells with totipotency features (2013)
Now, a dozen years after Yamanaka’s landmark discovery, the approach to cell and organ regeneration that seems to have a great deal of promise is more subtle but still involves OSKM. Instead of using the Yamanaka factors to regress cells to complete pluripotent status outside the body, the idea is to induce partial epigenetic regression of cells to younger states where they are in the body, where they still have memory of their cell lineage, and they happen to be already located in just the right places to induce renewal. A key to this line of research was the discovery that this form of renewal happens naturally all the time within the body. Naturally using the same Yamanaka factors. And further, that it is triggered by things we have traditionally regarded as bad, including cell senescence, inflammation and inflammasome’s. I spin the story out as I see it here, and state what I think the implications are for regenerative medicine and for the possibility of significantly longer lifespans. I repeat, this is basically a good news story.
Prelude
A few years ago it was becoming evident that a much more subtle approach needed to be taken to cell an organ regeneration them that tried earlier. The 2017 publication Transient transcription factor (OSKM) expression is key towards clinical translation of in vivo cell reprogramming points to the growing awareness back then of the need for a more subtle approach to OSKM administration, one that induces only a partial age regression in the cells that preserves their lineage memory. “Reprogramming adult, fully differentiated cells to pluripotency in vivo via Oct3/4, Sox2, Klf4 and c-Myc (OSKM) overexpression has proved feasible in various independent studies and could be used to induce tissue regeneration owing to the proliferative capacity and differentiation potential of the reprogrammed cells. However, a number of these reports have described the generation of teratomas caused by sustained reprogramming, which precludes the therapeutic translation of this technology. A recent study by the Izpisúa-Belmonte laboratory described a cyclic regime for short-term OSKM expression in vivo that prevents complete reprogramming to the pluripotent state as well as tumorigenesis. We comment here on this and other studies that provide evidence.”
Another 2017 publication Senescence-Inflammatory Regulation of Reparative Cellular Reprogramming in Aging and Cancer reported “– However, tissue rejuvenation should involve not only the transient epigenetic reprogramming of differentiated cells, but also the committed re-acquisition of the original or alternative committed cell fate. Chronic or unrestrained epigenetic plasticity would drive aging phenotypes by impairing the repair or the replacement of damaged cells; such uncontrolled phenomena of in vivo reprogramming might also generate cancer-like cellular states.” Or, in colloquial nativist English, grandpappy tells his young great-grandson: “When you grow up, doncha you go about fagettin wheah you came from.”
In-situ cell regeneration
The abstract of the 2018 publication In Vivo Transient and Partial Cell Reprogramming to Pluripotency as a Therapeutic Tool for Neurodegenerative Diseases provides a bit more complete summary of the situation: ”In theory, human diseases in which a specific cell type degenerates, such as neurodegenerative diseases, can be therapeutically addressed by replacement of the lost cells. The classical strategy for cell replacement is exogenous cell transplantation, but now, cell replacement can also be achieved with in situ reprogramming. Indeed, many of these disorders are age-dependent, and “rejuvenating” strategies based on cell epigenetic modifications are a possible approach to counteract disease progression. In this context, transient and/or partial reprogramming of adult somatic cells towards pluripotency can be a promising tool for neuroregeneration. Temporary and controlled in vivo overexpression of Yamanaka reprogramming factors (Oct3/4, Sox2, Klf4, and c-Myc (OSKM)) has been proven feasible in different experimental settings and could be employed to facilitate in situ tissue regeneration; this regeneration can be accomplished either by producing novel stem/precursor cells, without the challenges posed by exogenous cell transplantation, or by changing the epigenetic adult cell signature to the signature of a younger cell. The risk of this procedure resides in the possible lack of perfect control of the process, carrying a potential oncogenic or unexpected cell phenotype hazard. Recent studies have suggested that these limits can be overcome by a tightly controlled cyclic regimen of short-term OSKM expression in vivo that prevents full reprogramming to the pluripotent state and avoids both tumorigenesis and the presence of unwanted undifferentiated cells. On the other hand, this strategy can enhance tissue regeneration for therapeutic purposes in aging-related neurological diseases as well. These data could open the path to further research on the therapeutic potential of in vivo reprogramming in regenerative medicine.”_ There appears to be a great deal of excitement and ongoing research connected with this possibility, that is in vivo approaches to tissue regeneration.
(Following the usual custom in this blog, taking license as a blog writer, I sometimes give italic and/or bold emphasis of parts of quoted passages that I think are particularly important.)
One example of this current research is the July 2018 publication AAV vector-mediated in vivo reprogramming into pluripotency: “In vivo reprogramming of somatic cells into induced pluripotent stem cells (iPSC) holds vast potential for basic research and regenerative medicine. However, it remains hampered by a need for vectors to express reprogramming factors (Oct-3/4, Klf4, Sox2, c-Myc; OKSM) in selected organs. Here, we report OKSM delivery vectors based on pseudotyped Adeno-associated virus (AAV). Using the AAV-DJ capsid, we could robustly reprogram mouse embryonic fibroblasts with low vector doses. Swapping to AAV8 permitted to efficiently reprogram somatic cells in adult mice by intravenous vector delivery, evidenced by hepatic or extra-hepatic teratomas and iPSC in the blood. Notably, we accomplished full in vivo reprogramming without c-Myc. Most iPSC generated in vitro or in vivo showed transcriptionally silent, intronic or intergenic vector integration, likely reflecting the increased host genome accessibility during reprogramming. Our approach crucially advances in vivo reprogramming technology, and concurrently facilitates investigations into the mechanisms and consequences of AAV persistence.” While this publication is about introduction of OSKM into the body to trigger reprogramming, I think the more subtle, natural and interesting approach is that used in the body, such as the local triggering of OSKM by cell senescence and the inflammatory cytokine IL-6.
The bad is sometimes good
In terms of what goes on within the body, processes that we have traditionally associated with being “bad” appear to be essential for initiating regenerative processes which are ”good.” Specifically, cell damage, cell senescence, inflammation and the release of inflammatory cytokines are needed to initiate natural regenerative processes. In other words, the body automatically triggers natural regenerative processes when and where it senses they are needed, as outlined in the next two publication citations.
The 2016 publication Tissue damage and senescence provide critical signals for cellular reprogramming in vivo reports: “Reprogramming of differentiated cells into pluripotent cells can occur in vivo, but the mechanisms involved remain to be elucidated. Senescence is a cellular response to damage, characterized by abundant production of cytokines and other secreted factors that, together with the recruitment of inflammatory cells, result in tissue remodeling. Here, we show that in vivo expression of the reprogramming factors OCT4, SOX2, KLF4, and cMYC (OSKM) in mice leads to senescence and reprogramming, both coexisting in close proximity. Genetic and pharmacological analyses indicate that OSKM-induced senescence/ requires the Ink4a/Arf locus and, through the production of the cytokine interleukin-6, creates a permissive tissue environment for in vivo reprogramming. Biological conditions linked to senescence, such as tissue injury or aging, favor in vivo reprogramming by OSKM. These observations may be relevant for tissue repair.”
Another publication covering some of the same territory is the 2017 item Injury-Induced Senescence Enables In Vivo Reprogramming in Skeletal Muscle. “In vivo reprogramming is a promising approach for tissue regeneration in response to injury. Several examples of in vivo reprogramming have been reported in a variety of lineages, but some including skeletal muscle have so far proven refractory. Here, we show that acute and chronic injury enables transcription-factor-mediated reprogramming in skeletal muscle. Lineage tracing indicates that this response frequently originates from Pax7+ muscle stem cells. Injury is associated with accumulation of senescent cells, and advanced aging or local irradiation further enhanced in vivo reprogramming, while selective elimination of senescent cells reduced reprogramming efficiency. The effect of senescence appears to be, at least in part, due to the release of interleukin 6 (IL-6), suggesting a potential link with the senescence-associated secretory phenotype. Collectively, our findings highlight a beneficial paracrine effect of injury-induced senescence on cellular plasticity, which will be important for devising strategies for reprogramming-based tissue repair.”
Somatic body cells are much more plastic than we once thought
That is, they are much more susceptible to being epigenetically regressed and regenerated than anyone previously thought. “Breakthroughs in nuclear reprogramming have revealed that differentiated cells are strikingly plastic both in vitro and in vivo, with exciting implications for disease modeling and regenerative medicine (Srivastava and DeWitt, 2016, Takahashi and Yamanaka, 2016). Many organs, such as pancreas, liver, and kidney are permissive for in vivo reprogramming both to pluripotency and lineage switching (Abad et al., 2013, Srivastava and DeWitt, 2016), while other organs and tissues—most notably, skeletal muscle—do not develop teratomas. Interestingly, it has been shown that induced in vivo lineage reprogramming in liver and pancreas is more efficient when combined with injury (Heinrich et al., 2015). Moreover, transient induction of senescence occurs in non-muscle cells during regeneration following muscle damage (Le Roux et al., 2015). In light of these observations, we hypothesized that injury can promote reprogramming in vivo in skeletal muscle and that cellular senescence might play an important role during this process.(ref).”
Different cells in the same neighborhood can be doing very different things
A key thing pointed out here and amplified in other articles I cite below is that not all cells in a given neighborhood in an organ are doing the same thing, and in fact for cell regeneration to take place some have to be doing quite different things. That is some have to be injured or senescent or dying and releasing noxious cytokines in order for other cells in the neighborhood to be undergoing epigenetic regression and regeneration using the OSKM factors. Paracrine (in the neighborhood) Intercellular communication is required for this to take place. And, as the following article points out inflammatory cytokines are absolutely required for initiation of in vivo reprogramming. Sell and organ renewable involves an inter-cellular dance. That is, some cells have to be seriously injured and screaming for help for other neighboring cells to initiate a regeneration process
The April 2018 article Senescence promotes in vivo reprogramming through p16INK4a and IL-6 reports: “Cellular senescence is a damage response aimed to orchestrate tissue repair. We have recently reported that cellular senescence, through the paracrine release of interleukin-6 (IL6) and other soluble factors, strongly favors cellular reprogramming by Oct4, Sox2, Klf4, and c-Myc (OSKM) in nonsenescent cells. Indeed, activation of OSKM in mouse tissues triggers senescence in some cells and reprogramming in other cells, both processes occurring concomitantly and in close proximity. In this system, Ink4a/Arf-null tissues cannot undergo senescence, fail to produce IL6, and cannot reprogram efficiently; whereas p53-null tissues undergo extensive damage and senescence, produce high levels of IL6, and reprogram efficiently. Here, we have further explored the genetic determinants of in vivo reprogramming. We report that Ink4a, but not Arf, is necessary for OSKM-induced senescence and, thereby, for the paracrine stimulation of reprogramming. However, in the absence of p53, IL6 production and reprogramming become independent of Ink4a, as revealed by the analysis of Ink4a/Arf/p53 deficient mice. In the case of the cell cycle inhibitor p21, its protein levels are highly elevated upon OSKM activation in a p53-independent manner, and we show that p21-null tissues present increased levels of senescence, IL6, and reprogramming. We also report that Il6-mutant tissues are impaired in undergoing reprogramming, thus reinforcing the critical role of IL6 in reprogramming. Finally, young female mice present lower efficiency of in vivo reprogramming compared to male mice, and this gender difference disappears with aging, both observations being consistent with the known anti-inflammatory effect of estrogens. The current findings regarding the interplay between senescence and reprogramming may conceivably apply to other contexts of tissue damage.”
Again, we have the point that regeneration requires a dance between different cells in close proximity being in different states of distress or contentment. This last article also points out the good-and-bad possible roles of the OSKM factors themselves “The power of cellular senescence in inducing tissue remodelling has been further extended to processes of cellular reprogramming in vivo. The transgenic expression of the four transcription factors abbreviated as OSKM (Oct4, Sox2, Klf4, and c‐Myc) (Takahashi & Yamanaka, 2006) in adult mice induces dedifferentiation and cellular reprogramming within multiple tissues (Abad et al., 2013; Ohnishi et al., 2014). However, in addition to reprogramming, the activation of OSKM also results in cellular damage and senescence, both in vitro (Banito et al., 2009) and in vivo (Chiche et al., 2017; Mosteiro et al., 2016). Therefore, OSKM induces two opposite cellular fates, namely senescence and reprogramming, that coexist in vivo in separate, but proximal, subsets of cells (Chiche et al., 2017; Mosteiro et al., 2016). Importantly, it has been demonstrated that senescence plays an active role in facilitating in vivo reprogramming through the paracrine action of the SASP, being interleukin‐6 (IL6) a critical mediator (Chiche et al., 2017; Mosteiro et al., 2016). Of note, IL6 plays an important role also during in vitro reprogramming (Brady et al., 2013; Mosteiro et al., 2016). Moreover, the concept that senescence promotes cellular plasticity has been further extended to the activation of somatic stem/progenitor cells. In particular, the SASP can confer somatic stem/progenitor features onto proximal epithelial cells in several tissues (Ritschka et al., 2017).”
Portraying the SASP as an all-evil process that needs to be stamped out, as some researchers have done, is plum wrong
When things are working well in the body, the SASP is a normal part of the normal regeneration process.
The 2017 Ritschka article just mentioned is The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration which has to say: “Senescence is a form of cell cycle arrest induced by stress such as DNA damage and oncogenes. However, while arrested, senescent cells secrete a variety of proteins collectively known as the senescence-associated secretory phenotype (SASP), which can reinforce the arrest and induce senescence in a paracrine manner. However, the SASP has also been shown to favor embryonic development, wound healing, and even tumor growth, suggesting more complex physiological roles than currently understood. Here we uncover timely new functions of the SASP in promoting a proregenerative response through the induction of cell plasticity and stemness. We show that primary mouse keratinocytes transiently exposed to the SASP exhibit increased expression of stem cell markers and regenerative capacity in vivo. However, prolonged exposure to the SASP causes a subsequent cell-intrinsic senescence arrest to counter the continued regenerative stimuli. Finally, by inducing senescence in single cells in vivo in the liver, we demonstrate that this activates tissue-specific expression of stem cell markers. Together, this work uncovers a primary and beneficial role for the SASP in promoting cell plasticity and tissue regeneration and introduces the concept that transient therapeutic delivery of senescent cells could be harnessed to drive tissue regeneration.”
A caution about senolytic approaches to health and longevity
A key point in the last quote, also emphasized in many other articles, makes me question the wisdom of senolytic approaches advocated by some antiaging researchers, which is simply to extend health and life by getting rid of senescent cells. At best, these senolytic approaches by themselves may be too simplistic, and at worst they might be harmful by removing senescence triggers of cell rejuvenation. The garbage heap of dysfunctional therapies based on “getting rid of the bad stuff” already has many things heaped on it, including bloodletting to get rid of “bad humors” in the blood and colonic irrigation and enema therapies to get rid of “rotten matter in digestive track.” I wonder if senolytic therapies might be headed for the same garbage heap. On the other hand, if there is so much expression of the SASP so as to cause the above-mentioned cell-intrinsic senescence arrest, then conceivably a companion senolytic approach might be useful.
Also, while we are discussing the mixing up the biological good in the biological bad, we need to recall that the OSKM renewal factors are deeply involved in cancer processes, so much so that they are traditionally regarded to be carcinogenic.
In vivo regenerative programming can be used to transform one kind of cell to another related kind of cell
This is a point with many important implications, particularly related to the regeneration of non-mitotic cells such as brain neurons or hair stem cells which have traditionally been regarded to be irreplaceable. As a result, until very recently nerve regeneration in the brain has been regarded to be impossible.
However, the 2013 publication In vivo reprogramming reactive glia into iPSCs to produce new neurons in the cortex following traumatic brain injury pointed out the possibility of such regeneration and actually showed that it can be done in mouse brains. “Traumatic brain injury (TBI) results in a significant amount of cell death in the brain. Unfortunately, the adult mammalian brain possesses little regenerative potential following injury and little can be done to reverse the initial brain damage caused by trauma. Reprogramming adult cells to generate induced pluripotent stem cell (iPSCs) has opened new therapeutic opportunities to generate neurons in a non-neurogenic regions in the cortex. In this study we showed that retroviral mediated expression of four transcription factors, Oct4, Sox2, Klf4, and c-Myc, cooperatively reprogrammed reactive glial cells into iPSCs in the adult neocortex following TBI. These iPSCs further differentiated into a large number of neural stem cells, which further differentiated into neurons and glia in situ, and filled up the tissue cavity induced by TBI. The induced neurons showed a typical neuronal morphology with axon and dendrites, and exhibited action potential. Our results report an innovative technology to transform reactive glia into a large number of functional neurons in their natural environment of neocortex without embryo involvement and without the need to grow cells outside the body and then graft them back to the brain. Thus this technology offers hope for personalized regenerative cell therapies for repairing damaged brain.”
CELL REGENERATION AND CANCERS
There are a number of story threads connecting these topics, and I mentioned a few here.
The process of cell de-differentiation that characterizes the initial stage of cell regeneration may also play into the creation of cancers
The July 2018 publication Plasticity of differentiated cells in wound repair and tumorigenesis, part I: stomach and pancreas deals with this issue and the following diagram and legend are from there.
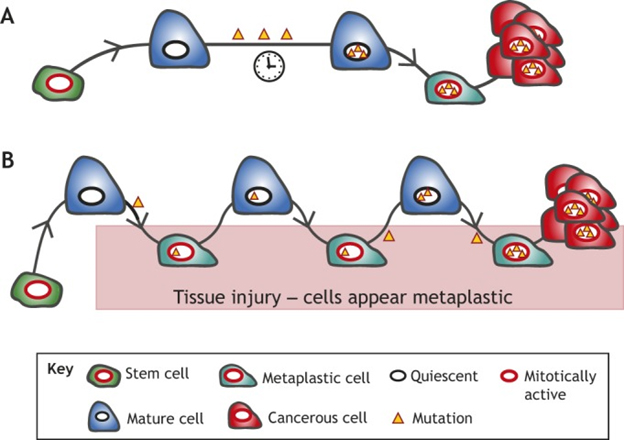
“Proposed models of mature cells acting as cancer cells of origin. We propose that long-lived mature cells may accumulate and store mutations, eventually acting as – or giving rise to cells that can act as – cells of origin for cancers in diverse tissues. This mutational accumulation may occur in two main ways: (A) mature cells (dark blue) may accumulate mutations (yellow triangles) as they maintain their mature functioning cell fate over time. The mutations themselves or stressors may trigger dedifferentiation (teal cell). If the acquired mutations are sufficiently carcinogenic, they may then block the cell in the dedifferentiated state, causing it to expand as a clone that can give rise to cancer (red). (B) The ‘cyclical hit’ model describes mature cells that dedifferentiate and redifferentiate multiple times in response to injury/inflammation. Each time the cells are called back into the cell cycle, replicative stress can promote mutation accumulation. Differentiated cells can store such mutations indefinitely. Eventually, a mutation or combination of mutations is sufficient to block the cell in one of its replicative phases and lead to clonal expansion and potential tumorigenesis.”
Note also the suggestion that a single cell may have a history of several cycle of de-differentiation and re-differentiation accumulating carcinogenic mutations on the way. I do not know the extent to which one or both of the models in the diagram importantly reflect what is going on. The cell renewal perspective provides a different way of looking at phenomena we thought we fairly well understood. Could it be the what we call cancer stem cells are actually somatic cells that have accumulated carcinogenic mutations and have been regressed to near stem cell status by the body’s natural application of the OSKM factors?
OSKM natural in-vivo de-differentiation may have a darker side: loss of cell lineage memory and epigenetic regulation may lead to cancers
This possibility is outlined in the late 2018 publication The causal relationship between epigenetic abnormality and cancer development: in vivo reprogramming and its future application. “There is increasing evidence that cancer cells acquire epigenetic abnormalities as well as genetic mutations during cancer initiation, maintenance, and progression. However, the role of epigenetic regulation in cancer development, especially at the organismal level, remains to be elucidated. Here, we describe the causative role of epigenetic abnormalities in cancer, referring to our in vivo studies using induced pluripotent stem cell technology. We first summarize epigenetic reorganization during cellular reprogramming and introduce our in vivo reprogramming system for investigating the impact of dedifferentiation-driven epigenetic disruption in cancer development. Accordingly, we propose that particular types of cancer, in which causative mutations are not often detectable, such as pediatric cancers like Wilms’ tumor, may develop mainly through alterations in epigenetic regulation triggered by dedifferentiation. Finally, we discuss issues that still remain to be resolved, and propose possible future applications of in vivo reprogramming to study cancer and other biological phenomena including organismal aging.”
Here is another2014 publication with the same theme Concise review: dedifferentiation meets cancer development: proof of concept for epigenetic cancer. “The technology for generation of induced pluripotent stem cells (iPSCs) has made significant contributions to various scientific fields, and the field of cancer biology is no exception. Although cancer is generally believed to develop through accumulation of multiple genetic mutations, there is increasing evidence that cancer cells also acquire epigenetic abnormalities during development, maintenance, and progression. Because the epigenetic status of somatic cells changes dynamically through reprogramming, iPSC technology can be utilized to actively and globally alter the epigenetic status of differentiated cells. Using this technology, a recent study has revealed that some types of cancer can develop mainly through disruption of the epigenetic status triggered by dedifferentiation. In this paper, we outline the reprograming process and the epigenetic mechanism associated with the maintenance or conversion of cell identity. We then describe several observations suggesting that dedifferentiation can play an important role in cancer development. Finally, we introduce the system responsible for in vivo reprogramming to demonstrate the involvement of dedifferentiation-driven epigenetic disruption in cancer development, and propose that particular types of cancer can develop predominantly through epigenetic alterations.”_
An interesting angle is that it may be possible to treat some cancers by in vivo reprogramming of cancer cells back to their original cell types
What a coup this could be if it worked! For example, glioblastoma is a rapid and sure-fire killer brain cancer for which there is no known therapy. As I write this there is widespread mourning for the loss of Senator John Mcain who just died of it. However a publication that appeared only a few days ago suggests a possible therapy: Reprogramming glioblastoma multiforme cells into neurons by protein kinase inhibitors. “Reprogramming of cancers into normal-like tissues is an innovative strategy for cancer treatment. Recent reports demonstrate that defined factors can reprogram cancer cells into pluripotent stem cells. Glioblastoma multiforme (GBM) is the most common and aggressive malignant brain tumor in humans. Despite multimodal therapy, the outcome for patients with GBM is still poor. Therefore, developing novel therapeutic strategy is a critical requirement. METHODS: We have developed a novel reprogramming method that uses a conceptually unique strategy for GBM treatment. We screened a kinase inhibitor library to find which candidate inhibitors under reprogramming condition can reprogram GBM cells into neurons. The induced neurons are identified whether functional and loss of tumorigenicity. RESULTS: We have found that mTOR and ROCK kinase inhibitors are sufficient to reprogram GBM cells into neural-like cells and “normal” neurons. The induced neurons expressed neuron-specific proteins, generated action potentials and neurotransmitter receptor-mediated currents. Genome-wide transcriptional analysis showed that the induced neurons had a profile different from GBM cells and were similar to that of control neurons induced by established methods. In vitro and in vivo tumorigenesis assays showed that induced neurons lost their proliferation ability and tumorigenicity. Moreover, reprogramming treatment with ROCK-mTOR inhibitors prevented GBM local recurrence in mice. CONCLUSION: This study indicates that ROCK and mTOR inhibitors-based reprogramming treatment prevents GBM local recurrence. Currently ROCK-mTOR inhibitors are used as anti-tumor drugs in patients, so this reprogramming strategy has significant potential to move rapidly toward clinical trials.”
Probably, most of the readers in this blog know about mTOR signaling and are familiar with some of the ways to inhibit or block it – e.g. metformin, resveratrol, rapamycin. But what is ROCK? The 2011 publication ROCKing Regeneration: Rho Kinase Inhibition as Molecular Target for Neurorestoration describes it. “Regenerative failure in the CNS largely depends on pronounced growth inhibitory signaling and reduced cellular survival after a lesion stimulus. One key mediator of growth inhibitory signaling is Rho-associated kinase (ROCK), which has been shown to modulate growth cone stability by regulation of actin dynamics. Recently, there is accumulating evidence the ROCK also plays a deleterious role for cellular survival. In this manuscript we illustrate that ROCK is involved in a variety of intracellular signaling pathways that comprise far more than those involved in neurite growth inhibition alone. Although ROCK function is currently studied in many different disease contexts, our review focuses on neurorestorative approaches in the CNS, especially in models of neurotrauma. Promising strategies to target ROCK by pharmacological small molecule inhibitors and RNAi approaches are evaluated for their outcome on regenerative growth and cellular protection both in preclinical and in clinical studies.”Regenerative failure in the CNS largely depends on pronounced growth inhibitory signaling and reduced cellular survival after a lesion stimulus. One key mediator of growth inhibitory signaling is Rho-associated kinase (ROCK), which has been shown to modulate growth cone stability by regulation of actin dynamics. Recently, there is accumulating evidence the ROCK also plays a deleterious role for cellular survival. In this manuscript we illustrate that ROCK is involved in a variety of intracellular signaling pathways that comprise far more than those involved in neurite growth inhibition alone. Although ROCK function is currently studied in many different disease contexts, our review focuses on neurorestorative approaches in the CNS, especially in models of neurotrauma. Promising strategies to target ROCK by pharmacological small molecule inhibitors and RNAi approaches are evaluated for their outcome on regenerative growth and cellular protection both in preclinical and in clinical studies.
Cell and tissue fate: locked-in inflammation? Cancer? Tissue and organ regeneration?
The same factors seem to be involved in a number of different situations with very different cell fate and health outcomes. These including cell senescence, NF-kB. IL-6, inflammation, the SASP and inflammasomes. To grapple with what happens when, some researchers suggest algorithmic threshold models. An example is the 2017 publication Senescence-Inflammatory Regulation of Reparative Cellular Reprogramming in Aging and Cancer. “– The degree of senescence/inflammation-associated deviation from the homeostatic state may delineate a type of thresholding algorithm distinguishing beneficial from deleterious effects of in vivoreprogramming. First, transient activation of NF-κB-related innate immunity and senescence-associated inflammatory components (e.g., IL-6) might facilitate reparative cellular reprogramming in response to acute inflammatory events. Second, para-inflammation switches might promote long-lasting but reversible refractoriness to reparative cellular reprogramming. Third, chronic senescence-associated inflammatory signaling might lock cells in highly plastic epigenetic states disabled for reparative differentiation. The consideration of a cellular reprogramming-centered view of epigenetic plasticity as a fundamental element of a tissue’s capacity to undergo successful repair, aging degeneration or malignant transformation should provide challenging stochastic insights into the current deterministic genetic paradigm for most chronic diseases, thereby increasing the spectrum of therapeutic approaches for physiological aging and cancer.”
The following diagrams and explanations are from that publication.
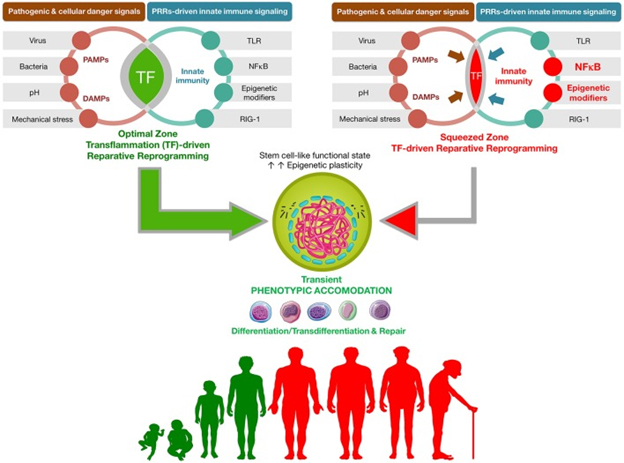
“Transflammation-driven epigenetic plasticity: a paradigmatic example of in vivo reparative reprogramming. Transient activation of the PAMPs-DAMPs → NFκB signaling axis may delineate an optimal zone of transflammation (TF)-driven reparative reprogramming characterized by increased epigenetic plasticity and phenotypic malleability capable of responding and adapting to injury, stress, and disease (Lee et al., 2012; O’Neill, 2012; Cooke et al., 2014). The efficiency of NFκB signaling and the level of inflammatory responses is the nodal point linking the pathogenic assault and cellular danger signals and the organization of cellular resistance and tissue repair. NFκB hyperfunction and its interaction with epigenetic modifiers would significantly squeeze the optimal zone of TF-driven reparative reprogramming, thus impairing the adequate organization of defense mechanisms. By operating as the perpetrator of inflammaging, the NFκB signaling integrates the intracellular regulation of transflammation immune responses in both aging and aging-related diseases (Salminen et al., 2008; Montgomery and Shaw, 2015).”
This diagram is interesting because it suggests an additional role for NF-kB signaling beyond that of initiation and perpetration of inflammatory responses, which I have discussed in a number of past blog entries in the inflammation series. As I understand it, the “squeezed zone” in the diagram corresponds to a situation of chronic inflammation and the over activation of NFκB signaling characteristic of aging which essentially inhibits cellular transdifferentiation and repair. As suggested by the red figures in the lower part of the diagram which are aging people whose cellular renewal mechanisms are inhibited by their chronic inflammation. Or, simply put, chronic inflammation stops reparative reprogramming.
If this hypothesis is correct it is exceedingly exciting to me on both highly personal and professional levels. This is because I think I know how to create an effective at least partial bulwark against chronic inflammation, and that is the 4 Herb Synergy liposomal concoction I have been consuming for at least four years now and soon will be making available for others to use. (A blog entry on this is coming very soon now.) Could it be that with my age now approaching 89 in November, my ability to control chronic inflammation has been a major factor in the preservation of my health and avoidance of any of the chronic diseases characteristic of advanced aging, precisely because it has preserved my body’s capability for self renewal? Further, is there something here I could share with others interests of their health? Or are there other key factors beyond those considered here which have to be taken into account?
Here is another diagram from the same publication related to the same options of effective reparative reprogramming versus pathological reprogramming:
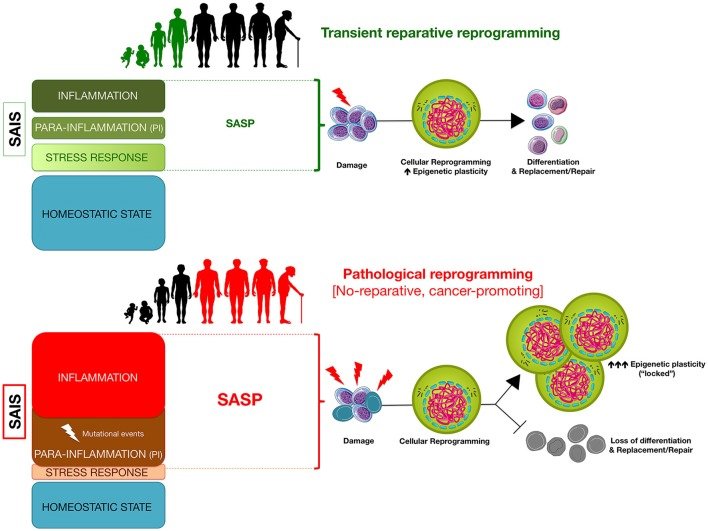
“Senescence-associated inflammatory signaling (SAIS)-regulated in vivo reprogramming: a threshold model of epigenetic plasticity in aging and cancer. The degree of senescence/inflammation deviation from the homeostatic state delineates a thresholding algorithm distinguishing beneficial vs. deleterious effects of in vivo reprogramming. First, transient activation of innate immunity and/or SASP components (e.g., IL-6) might facilitate reparative cellular reprogramming in response to acute inflammatory events. Second, NFκB-dependent and NFκB-independent (e.g., SIR) para-inflammation switches might promote a long-lasting but reversible refractoriness to reparative cellular reprogramming. Third, chronic SASP might lock cells into highly plastic epigenetic states disabled for reparative differentiation capacities.”
Again, this diagram suggests that control chronic inflammation may make the difference between the states identified in the upper lower part of the diagram. It suggests that by controlling and minimizing chronic inflammation we could possibly be able to maintain a condition where transient reparative reprogramming is effective throughout life. I do not know what this would mean for longevity, but the news would be very good.
Also, recall the passage cited above: “However, prolonged exposure to the SASP causes a subsequent cell-intrinsic senescence arrest to counter the continued regenerative stimuli.(ref). So effective cell regeneration requires some but not too much of the SASP.” Here is where senolytic approaches could be valuable.
A further suggestion to me at this point is that maintenance of high levels of NAD+ and Sirtuins might also be required for transient reparative reprogramming to be effective. On a personal level I do this by supplementation with resveratrol and nicotinamide riboside to help maintain both NAD+ and Sirtuin levels.
A third diagram from the same publication follows:
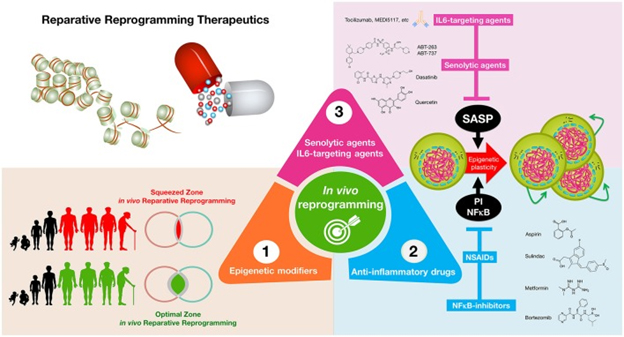
“Reparative reprogramming therapeutics: enhancing the body’s self-cell therapy for resistance to damage and disease. A cellular reprogramming-centered view of epigenetic plasticity as a fundamental dimension of a tissue’s capacity to undergo successful repair may provide new therapeutic approaches for aging and cancer. (1) Epigenetic modifiers: small molecules capable of mimicking the transient amelioration of tissue functions occurring upon short-term induction of OSKM-induced nuclear reprogramming (Mahmoudi and Brunet, 2016; Ocampo et al., 2016) might increase epigenetic plasticity and to enhance regeneration in aging tissues; (2) anti-inflammatory drugs: NFκB-targeting drugs and commonly employed NSAIDs might help reduce some aging- and cancer-promoting inflammatory feedback loops to reestablish the functioning of reparative reprogramming; (3) IL-6-targeting and senolytic agents: IL-6 blockade and senescent cell ablation might help unlock the chronic epigenetic plasticity of SASP-damaged tissues to successfully achieve tissue rejuvenation if accompanied by reparative differentiation phenomena.”
MSI-1436 – a related story about regeneration
From when I first started seriously studying biology and writing this blog, I would come across research articles about how certain organisms could regenerate part of themselves, like newts, tadpoles and hydras. But back then we had only vague ideas of how to induce this kind of regeneration and complex mammals like ourselves. This situation is now changed and there is a developing literature of approaches to regeneration that can apparently work. One of the stories is about a substance called MSI-1436.
The 2017 publication The protein tyrosine phosphatase 1B inhibitor MSI-1436 stimulates regeneration of heart and multiple other tissues reports: “Regenerative medicine holds substantial promise for repairing or replacing tissues and organs damaged by disease, injury, and degeneration. Much of the field has focused on development of cell-based therapeutics, gene-based therapeutics, and tissue engineering-based therapeutics. In contrast, development of small molecule regenerative medicine therapies is an emerging area. Using the adult zebrafish as a novel screening platform, we identified MSI-1436 as a first-in-class regenerative medicine drug candidate. MSI-1436 is a naturally occurring aminosterol that inhibits protein tyrosine phosphatase 1B. Treatment of adult zebrafish by intraperitoneal injection of MSI-1436 increased the rate of regeneration of the amputated caudal fin, which is comprised of bone, connective, skin, vascular and nervous tissues and also increased the rate of adult zebrafish heart regeneration. Intraperitoneal administration of MSI-1436 to adult mice for 4 weeks after induction of myocardial infarction increased survival, improved heart function, reduced infarct size, reduced ventricular wall thinning and increased cardiomyocyte proliferation. Satellite cell activation in injured mouse skeletal muscle was stimulated by MSI-1436. MSI-1436 was well tolerated by patients in Phase 1 and 1b obesity and type 2 diabetes clinical trials. Doses effective at stimulating regeneration are 5–50-times lower than the maximum well tolerated human dose. The demonstrated safety and well established pharmacological properties of MSI-1436 underscore the potential of this molecule as a novel treatment for heart attack and multiple other degenerative diseases.”
Further, MSI-1436 has already been in clinical trials for other specific indications and is known to be safe for human administration. “MSI-1436 was well tolerated by patients in Phase 1 and 1b obesity and type 2 diabetes clinical trials. Doses effective at stimulating regeneration are 5–50-times lower than the maximum well tolerated human dose. The demonstrated safety and well established pharmacological properties of MSI-1436 underscore the potential of this molecule as a novel treatment for heart attack and multiple other degenerative diseases.” Demonstrated safety could shave many years off the time for this drug to be available as a mainline therapy. And its role as a drug could be very significant
MSI-1436 is a drug candidate for restoration of heart muscle function following a heart attack. Currently, no drug exists to restore heart muscle function after a heart attack. Cardiovascular disease is the world’s leading killer, taking the lives of around 18 million people every year, according to the World Health Organization, and disabling millions more. Imagine your cardiologist telling you “Yeah, your heart tissues are seriously compromised. However, all we have to do is regenerate some of those tissues, and you will be fine. No operation necessary. The process is noninvasive.” Clearly though, it will be sometime before the drug clears all the hurdles necessary to get to be available on the market.
Preserving umbilical cord blood – another approach to immune system regeneration
The June 2018 publication The potential of non-myeloablative heterochronous autologous hematopoietic stem cell transplantation for extending a healthy life span reports: “Aging is a complex multifactorial process, a prominent component being the senescence of the immune system. Consequently, immune-related diseases develop, including atherosclerosis, cancer, and life-threatening infections, which impact on health and longevity. Rejuvenating the aged immune system could mitigate these diseases, thereby contributing to longevity and health. Currently, an appealing option for rejuvenating the immune system is heterochronous autologous hematopoietic stem cell transplantation (haHSCT), where healthy autologous bone marrow/peripheral blood stem cells are collected during the youth of an individual, cryopreserved, and re-infused when he or she has reached an older age. After infusion, young hematopoietic stem cells can reconstitute the compromised immune system and improve immune function. Several studies using animal models have achieved substantial extension of the life span of animals treated with haHSCT. Therefore, haHSCT could be regarded as a potential procedure for preventing age-related immune defects and extending healthy longevity. In this review, the pros, cons, and future feasibility of this approach are discussed.”
Implications for anti-aging therapies
I could go on and make this writing two or three times as long, so much is the relevant research. I will finish, however, with some additional review articles citations showing excitement among aging researchers, and pick the central theme up again in a later blog entry.
Elixir of Life: Thwarting Aging With Regenerative Reprogramming. 2018
Anti-Aging Strategies Based on Cellular Reprogramming. 2016
Cellular reprogramming: A new way to understand aging mechanisms. 2018
Programming and Reprogramming Cellular Age in the Era of Induced Pluripotency. 2015
Changes in Regenerative Capacity through Lifespan. 2015
Rejuvenation by Partial Reprogramming of the Epigenome. 2017
iPSCs-based anti-aging therapies: Recent discoveries and future challenges. 2016
Direct cardiac reprogramming: progress and challenges in basic biology and clinical applications. 2015
Cardiomyocyte generation using stem cells and directly reprogrammed cells. 2012
Biology of Healthy Aging and Longevity. 2016
Mitochondrial function in pluripotent stem cells and cellular reprogramming. 2014

Thanks Vince, Nice analysis.
As the regeneration process is an high energy-demanding process, do you see any link with the mitochondria status?
Vince, have you looked into photobiomodulation, and if so, what are your thoughts on it?
What is the status of your 4 herb liposomal synergistic brew? I gather curcumin and ashwaganda, what are the other two?
Sorry for the delay in answering. We have product in bottles and our selling website should be up in just a few days now. Check the latest blog entries with extensive info on the supplement (reply on 6/10/19)
The other two herbs are ginger and boswellia, actually extracts of them enhanced for the bioactive ingredients. Should be sold in a few days now, as of 6/10/19
Vince
The other two are ginger and Boswellia. See the 2019 blog entries The making of a dietary supplement – the long and short histories of it, And INFLAMMATION PART 6: THE SCIENCE BEHIND THE 4 HERB SYNERGY DIETARY SUPPLEMENT.
Vince
Pingback: INFLAMMATION PART 6: THE SCIENCE BEHIND THE 4 HERB SYNERGY DIETARY SUPPLEMENT - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™
Pingback: FUNNY THINGS ARE HAPPENING TO ME ON THE WAY TO 100 - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™
Pingback: CHRONOBIOLOGY OF TISSUE REGENERATION - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™
Pingback: YOUNGING1– THE EMERGING AGING REVERSAL STRATEGY - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™