
By James P Watson with comments and editorial assistance by Vince Giuliano
Introduction by Vince Giuliano:
This is the third blog entry in the series NAD+ an emerging framework for health and life extension. The first two entries were Part 1: The NAD World, and Part 2: Deeper into the NAD World, hopeful interventions We invite our readers to review these for background. The Part 4 blog entry in the NAD World series is concerned with the NQ01 gene, the Warburg effect, SIRT 1 and inflammation, and possible interventions. The Part 5 blog entry in the series is concerned with the conflicting roles of extra-cellular NAMPT, which behaves as both an enzyme and a cytokine, how it is implicated in inducing many disease processes as well as having some positive health impacts.
Ever since David Sinclair’s seminal 2013 publication relating insufficiency of NAD+ to mitochondrial dysfunctionality(ref) , and and for some time before that, many research scientists have thought that insufficient nuclear levels of NAD+ are a major cause of diseases and aging processes including mitochondrial dysfunctionality, associated deleterious metabolic consequences, and insufficient DNA repair. Based on much research, this point seems to be solid and incontrovertible. Having higher levels of NAD+ would doubtlesly be a good thing, especially for older people whose levels decline with age. The major though not the only avenues of benefit are realized via sirtuins, particularly SIRT1. As is well-recognized and pointed out in many of out earlier blog entries, the sirtuins are essential for multiple key biological processes such as DNA repair and healthy mitochondrial metabolism. Further, with aging and disease they tend to be in declining supply. When there is not enough SIRT1. serious consequences can ensue .
In the growing excitement about NAD science during last two years, some researchers and entrepreneurs have further thought that human NAD+ levels can likely be enhanced by ongoing supplementation with NAD precursors such as nicotinamide riboside (NR) or with nicotinamide mononucleotide (NMN), The thinking has been that such supplementation might make a significant difference in human health and longevity. However direct evidence that NAD levels can be non-transiently enhanced in humans, either intracellular or extra-cellular, is thin to nonexistent. No direct research shows this. Likewise, evidence that human health or longevity benefits will result from continuous NAD precursor supplementats is equally thin, Enhancement of SIRT1 levels in mice via NMN supplementation and associated health benefits has been observed in only short term trials with mice, ones that lasted only 7 or 10 days (ref)(ref). There appears to be no clear evidence however, either human or animal, that continuing to take a NAD precursor supplement over a long term can lead to higher continuing levels of NAD+ or the many health benefits hypothesized to ensue.
This has led Jim Watson to investigate just what determines human NAD levels and what determines SIRT1 levels. He found at least 50 such factors In this current blog entry reports on what he believes are the most important 30 of these.
This is another Chapter in the extended NAD story, but not the final one. We expect soon to publish a Part 4 blog entry in this NAD World series. That blog entry will look with more detail into several additional areas including NAD/Sirtuins and inflammation, and NAD and the much-discussed Warburg effect. Despite a now-popular perception that scarcity of NAD+ is the main cause of the Warburg effect, Jim Watson points out that the effect has multiple other causes including three long non-coding RNAs that promote it happening as a key effect in aging and cancer processes. Related to these causes,Jim discusses some possible intervention strategies that go beyond those normally discussed in the longevity literature.
1. Subcellular compartment levels of NAD regulate aging
The content of NAD in the nucleus as well as total cellular NAD levels declines with aging.
A. Animal studies that showed how total cellular NAD decline with aging – DNA damage seems to be the most significant biomarker that correlates with the change in cellular NAD levels
Several papers in the 1990s showed that PARP activity increased with aging in both animal and human cells, but the evidence that NAD declined with aging was not discovered until the past 10 years. In fact, a 1983 paper by Chapman, Zaun, and Gracy suggested the opposite effect – that NAD levels increased with aging. The first paper to show that NAD depletion occurred was actually a heart failure study published in 2005. Pillai and colleagues showed that PARP1 activation resulted in NAD depletion, reduced SIRT2 activity, and myocyte cell death. The next report was in 2008 by Parihar and colleagues, and was a study of rat hippocampal neurons. In this in vitro study, they showed that a 50% decline in NAD(H) levels occurred in the aged neurons. (see reference below). The 3rd report was a study in Wistar Rats, which showed a 4-7 fold decline in NAD+ levels with aging. ThIs paper by Braidy and colleagues at the U of New South Wales in Sidney, Australia was the first study that specifically was designed to look at declining levels of NAD with aging. (2011). What was remarkable in this study was that the DNA damage biomarker, pH2AX, seemed to correlate with the decline in NAD levels, even more than other biomarkers for oxidative stress.
B, Human Studies showing that total cellular NAD levels decline with aging – DNA damage seems to be the most significant biomarker that correlates with the change in cellular NAD levels
Until recently, it was not known if human cells also underwent a decline in NAD content with aging. The first paper to show this effect in humans was also from Sidney, Australia. In 2012, Massudi and colleagues showed a precipitous drop in NAD levels within human skin cells, harvested from areas of skin with no sun exposure from infancy to old age. Although measures of oxidative stress changed with aging (TBARS, MDA, F2-isoprostanes, etc.), the most significant change that correlated with aging was the DNA damage marker, pH2AX, which showed a significant increase with age in both males and females (p < 0.003). This suggests that the most important feature of cellular aging is DNA damage, not lipid oxidation or oxidative stress, per se. Along with this increase in DNA damage was a dramatic increase in PARP activity (10-fold) in males. PARP enzymes are NAD-consuming enzymes that sense and help repair single stranded DNA breaks via the base-excision repair (BER) pathway. Paradoxically, the increase in PARP activity was not statistically significant in the female cohort. Even more surprising was the fact that SIRT1 activity did now show a statistically significant change with aging in either males or females, however. Again, this is puzzling, since NAD levels declined so much.
References:
- · Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats
- · Age-associated changes in oxidative stress and NAD+ metabolism in human tissue
C, Studies that showed how nuclear levels of NAD decline with aging – Studies of Wallerian degeneration in the brain and mitochondrial biogenesis in muscles linked NAD deficiency in the nuclear subcellular compartment to the occurrence of Wallerian degeneration, and the “mitochondrial failure” seen with aging.
Long ago, there was circumstantial evidence that NAD levels may be regulated on a subcellular compartmental level, the significance of this was unknown, since no one had measured NAD levels in various sub cellular compartments. For instance, it was well-known that three isoforms of NMNAT existed [NMNAT-1 in nuclear compartment, NMNAT-2 in the Golgi complex, and NMNAT-3 in the mitochondria], the significance of this was not understood. NMNAT is a key enzyme since it is the only enzyme shared by both pathways for NAD synthesis, the NAD biosynthesis de novo pathway and the NAD Salvage cycle pathway.
One clue to this significance was the discovery of a mutant mouse model of Wallerian degeneration called the “Wlds mouse“, which was protected from developing Wallerian degeneration following axonal injury. It was discovered that the Wlds mouse had an 3 extra copies of a 85 kb tandem triplication on the end of chromosome 4 that contained two protein-coding genes for UCH-L1 (UPS gene) and NMNAT-1 (NAD synthesis gene for the nuclear isoform of NMNAT). These extra copies of the NMNAT-1 gene in the Wlds mouse resulted in a 3-fold higher NMNAT activity in the brain, but whole brain NAD+ levels were not increased. This Wlds mouse study was published by Wang and He from Boston in 2009, but they did not show that nuclear NAD levels were actually reduced with aging or Wallerian degeneration.
More recently, in a December 2013 paper Gomes and colleagues from Sinclair’s lab in Boston showed that in mice, declining NAD levels in the nucleus of muscle cells induced a pseudohypoxic state, disrupting nuclear-to-mitochondrial communication with aging. In 3 separate experiments, he “knocked down” NMNAT-1, NMNAT-2 and NMNAT-3. Only NMNAT-1 knock-down mimicked aging and the mitochondrial dysfunction seen with aging. Although not published, Sinclair has measured NAD levels in the nuclear subcellular compartment and has showed that nuclear NAD levels decline with aging in mice. In his key 2013 paper, he did publish evidence that IP supplementation with 400 mg/kg of NMN for one week reversed this “pseudohypoxic state” by promoting Tfam-dependent transcription of mitochondrially-encoded OXPHOS genes.
In summary, the indirect evidence about nuclear NAD levels from Wang and He’s work with the Wlds mice with 3 extra copies of the nuclear-specific isoform of NMNAT (NMNAT-1) as well as the more direct work on NMNAT-1 knockdown by Gomes and colleagues in Sinclair’s laboratory have provided tantalizing clues to a major cause of a “Universal phenotype of Aging:” that of the decline in mitochondrial-encoded OXOPHOS genes, which includes increased ROS generation, Warbug-type metabolism, oxidative stress, NAD depletion, and eventually ATP depletion in cells. By turning on the Tfam gene via a SIRT1-mediated mechanism, induced by intraperitoneal high-dose NMN supplementation, Gomes and colleagues increased expression of mitochondrial DNA encoded genes, thereby increasing the expression of mitochondrially-encoded OXPHOS genes.
Caveat: The above three paragraphs make a strong case that one major cause of aging is a decrease in the nuclear levels of NAD co-enzyme, required for so many nuclear proteins involving DNA repair, epigenetic gene regulation, and apoptosis. Unfortunately, no one has yet pin-pointed the cause of this “nuclear NAD decline” or demonstrated that it can be reversed for more than one week. The rest of this blog entry goes over 29 possible reasons why nuclear levels of NAD decline with aging. Until someone demonstrates that they can permanently restore nuclear NAD levels, the exact cause of this aging phenomena must still be considered speculative. Koch’s postulates must be proven.
2. NAD/NADH ratio regulates aging independently of NAD content
–The longevity gene, NQO1, regulates aging by altering the NAD/NADH ratio in cells. NQO1 does this by oxidizing NADH to NAD. Beta-lapachone increases NQO1 enzyme activity and quercetin increases Nrf2-mediated gene expression of NQO1.
Not only is the NAD content in the nucleus important for delaying/preventing aging, the redox ratio of NAD/NADH is also very important for delaying/preventing aging independently of the total NAD found in the cell. Many genetic studies in model organisms have searched for “longevity genes” that regulate lifespan. One of the curious findings from these studies is the gene that codes for the protein “NADH-quinone oxidoreductase 1″, or NQO1. NQO1 oxidizes NADH to NAD, thereby increasing the NAD/NADH ratio. Interestingly, Lee and colleagues from Korea recently showed that feeding animals beta-lapachone (aka Beta-L), an exogenous NQO1 co-substrate, prevented the age-dependent decline of motor and cognitive function in aged mice. Beta-lapchone is a compound originally obtained from the Lapcho tree and has been used for medical purposes for many years. Beta-L fed mice did not alter their food intake or locomotor activity, but did increase their energy expenditure as measured by VO2max and by heat generation. The Beta-L fed mice developed changes in gene expression that mimicked 30% caloric restricted diets. Another molecular effect of beta-Lapachone is that it induces apoptosis in breast and prostate cancer cells.
Gene polymorphisms in the NQO1 gene are strong prognostic indicators for breast cancer. For instance, the NQO1 2 genotype (P187S) predicts poor survival from breast cancer(ref). The relative risk for breast cancer in this with the P187S genotype is 6.15, when compared to control groups. The P187S genotype does not affect local recurrence, but affects survival.
Interestingly, benzene poisoning is associated with the mutation of the NQO1 gene at codon 187, which creates the 609C-T mutation of the NQO1 gene. This results in complete loss of the enzymatic activity of NQO1 protein. By this mechanism, benzene produces a NOA1 “loss-of-function” mutation and induces hematological malignancies. This appears to be a major mechanism for chemotherapy-induced secondary malignancies, which are called “therapy-related malignancies”. Two diseases that benzene induces are “therapy-related leukemia” and “therapy-related myelodysplastic syndrome“. The NQO1 gene can also be transcriptionally unregulated by a polyphenol called quercetin. Specifically, quercetin increases gene expression of the NQO1 gene via an Nrf2 transcription factor mediated pathway(ref)(ref)(ref). Specfically, quercetin enhances the binding of Nrf2 to the NRF-ARE binding site on the NQO1 gene promoter. Quercetin also increases Nrf2-mediated transcriptional activity by up regulation gut e expression of Nrf2 mRNA and Nrf2 protein. Quercetin also reduces the level of Keap1 protein, the binding partner of Nrf2, which prevents Nrf2 nuclear translocation. Quercetin reduces Keap-1in a post-translational mechanism, thereby reducing Nrf2 ubiquitination and proteasomal degradation. Another unusual up regulator of NQO1 is the toxin, dioxin.
The NAD World Part 4 blog enry contains additional discussion related to NQ01 and how it is egulated, beta lapachone and other topics mentioned in this item
References:
- LongevityMap Gene – Gene details HGNC symbol NQO1
- Nrf2 mediates cancer protection but not prolongevity induced by caloric restriction
- The Protein Level of PGC-1α, a Key Metabolic Regulator, Is Controlled by NADH-NQO1
- In Vivo Role of NAD(P)H:Quinone Oxidoreductase 1 (NQO1) in the Regulation of Intracellular Redox State and Accumulation of Abdominal Adipose Tissue
- Pharmacological activation of NQO1 increases NAD⁺ levels and attenuates cisplatin-mediated acute kidney injury in mice
- Enhanced activation of NAD(P)H: quinone oxidoreductase 1 attenuates spontaneous hypertension by improvement of endothelial nitric oxide synthase coupling via tumor suppressor kinase liver kinase B1/adenosine 5′-monophosphate-activated protein kinase-mediated guanosine 5′-triphosphate cyclohydrolase 1 preservation
- β-Lapachone ameliorates murine cisplatin nephrotoxicity: NAD+, NQO1, and SIRT1 at the crossroads of metabolism, injury, and inflammation
3. Clock/BMAL1
CLOCK and BMAL1 regulate the circadian expression of the SIRT1 gene. Day/night cycles are thus the #1 factor that determines SIRT1 expression.SIRT1 regulates circadian gene expression by deacetylating PER2
With long lists like those in this blog entry, it is easy to throw up your hands and say “its too complicated!” Well it isn’t! If you want to skip the list and just ask “What is the most important regulator of SIRT1 expression,” I do not think anyone would argue with the statement that CLOCK/BMAL1 bind to the gene promoter for SIRT1 and regulate the diurnal change in SIRT1 gene expression. (see references).
For example, it has been well-documented that liver insulin sensitivity correlates with the two circadian transcription factors CLOCK and BMAL1. BMAL1, CLOCK, and SIRT1 all must work together to “turn on” and “turn off” 15% of the genome in human cells every day. Unless the expression of these three proteins is coordinated, hepatic insulin resistance develops. Constant darkness “dysregulates” the coordinated timing of BMAL1 and SIRT1. As a result, BMAL1 and SIRT1 expression decreases with constant darkness and hepatic insulin resistance is induced. Interestingly resveratrol can dramatically reverse the “dysregulation” of SIRT1-dependent circadian genes by increasing SIRT1 activity. SIRT1 regulates circadian gene expression by PER2 deacetylation.
References:
- CLOCK/BMAL1 regulates circadian change of mouse hepatic insulin sensitivity by SIRT1
- The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control
- SIRT1 regulates circadian clock gene expression through PER2 deacetylation
- Interaction of circadian clock proteins PER2 and CRY with BMAL1 and CLOCK
4. Long non coding RNAs
The long non-coding RNA transcribed from the anti-sense strand of the SIRT1 gene regulates SIRT1 gene expression via a “trans-regulatory” mechaism. Long non-coding RNAs also appear to regulate cellular senescence, metabolism and many other cellular functions.
Introduction/Background
Recently, there has been dramatic shifts in the thinking of geneticists about the role of “junk DNA” in human disease, aging, and normal development. Specifically, over 56,000 unique RNA transcripts have been identified via next generation sequencing (also called “deep sequencing” or RNAseq). Of these, over 9,000 long non-coding RNA “genes” have been identified that make many more than 9,000 different RNA transcripts. Although there are many other types of non coding RNA besides long non-coding RNA, these RNA sequences that are > 200 base pairs in length have garnered the most attention since they seem to have such powerful regulatory functions over and above even epigenetic gene regulatory mechanisms.
Some recent papers have suggested that of the 6,000+ gene polymorphisms (SNPs) that have been linked to disease by GWAS studies, as many as 93% of these SNPs are not due to protein-coding regions of the human genome, but instead are in regulatory areas. Another surprising finding was that at least 5,000 (of the more than 9,000 DNA sequences that encode for long non-coding RNA) are not evolutionarily conserved. What does this mean? It means that these long non-coding RNA may be the “youngest” forms of gene regulation and may account for the unique characteristics of homo sapiens, such as our ability to make tools, form language, develop written communication, music, art, religion, socialization, etc. In short, long non-coding RNA may be why we are “human”, rather than looking and acting more like our nearest cousins, the chimpanzees and bonabos.
SIRT1 gene-specific lncRNA
It is not surprising then that a long non-coding RNA has been discovered that regulates SIRT1 gene expression. (see reference below). This long non-coding RNA is transcribed from the anti-sense strand of the SIRT1 gene. This lncRNA is called “SIRT1 antisense long non coding RNA” or SIRT1AS lncRNA for short. Wang and colleagues from Shanghai, China discovered this and published their finding in April, 2014. They were able to show that SIRT1 AS lncRNA expression results in an increase in expression of the SIRT1 gene. They isolated SIRT1 AS lncRNA from differentiating myotubes from developing skeletal muscle, as well as from the spleen. It appears that the SIRT1 AS lncRNA expression is both temporal and tissue-specific. For instance, higher levels of SIRT1 AS lncRNA were expressed in undifferentiated, younger tissues, Likewise, SIRT1 AS lncRNA was expressed in a tissue-specific fashion. In the spleen, SIRT1 AS lncRNA levels were higher than in skeletal muscle.
SIRT1 AS lncRNA may counteract microRNA that inhibit SIRT1
miRNA that down regulate SIRT1 gene expression appear to be blocked or their effect attenuated by the SIRT1 AS lncRNA. Thus, the function of SIRT1 AS lncRNA may be to counteract the effects of miR-34a, miR-217, miR-181a, and the other microRNA that increase SIRT1 mRNA degradation and thereby reduce SIRT1 protein expression.
Long non coding RNAs and cellular senescence
Gene-specific lncRNAs can regulated only one specific gene, like the SIRT1 AS lncRNA described above. However, many more lncRNAs regulate many genes rather than one specific gene. Gene-specific lncRNAs typically regulate nearby genes, which is a method called “regulation in-cis”. Since SIRT1 plays a role in cellular senescence, it is likely that the “Anti-sense” senescence-associated lncRNAs play an role in regulating genes that SIRT1also regulates. A list of the top 12 up-regulated Antisense lncRNAs with cellular senescence along with a list of the top 15 Anti-sense lncRNAs that are down regulated with cellular senescence are listed below.
In addition to the Anti-sense lncRNAs described above, a number of lncRNAs have been found that originate in pseudogenes, genes that no longer code for proteins, but are still transcribed from their sense or anti-sense strand. lncRNAs can regulate a single gene or regulate hundreds of genes as well. lncRNAs that regulate many genes typically do so via a method called “regulation in-trans”. These lncRNAs are not located near by the genes that they regulate, but instead, are typically located long distances away, even on other chromosomes. Many of these long non-coding RNAs are found in areas with no nearby protein-coding genes. For this reason, they are called “Long intergenic non coding RNAs” or lincRNAs.
Several lincRNAs have been discovered that regulate cellular senescence via “trans regulatory” methods. These lncRNAs appear to be “novel” and not within or associated with a particular protein-coding gene or a pseudogene.
Long non coding RNAs and metabolism
Since lncRNAs have been discovered that regulate cellular senescence, it is no surprise to find out that lncRNAs regulate metabolism as well. There is strong evidence that insulin resistance may be mediated in part by lncRNAs. This includes tissue-specifc lncRNAs and generalized lncRNAs. For instance, the long non coding RNA called “H19″ may be involved in the intergenerational transmission of diabetes mellitus. In a large next-generation RNA sequencing study of pancreatic RNA transcripts, over 1,000 lncRNAs were discovered in pancreatic islet beta cells. Of these, 40% were long intergenic non coding RNAs (lincRNAs) and 55% were Anti-sense strand long non-coding RNAs (AS lncRNAs). Interestingly, the non-coding RNA sequences found via this next generation sequencing study were more “tissue-specific” than the mRNA for protein-coding genes in the pancreatic tissue examined. In other words, non-protein coding RNAs had a more “tissue specific signature” than the protein-coding mRNAs that were sequenced. This is quite remarkable.
References:
- 2014 Identification, stability and expression of Sirt1 antisense long non-coding RNA
- 2013 Senescence-associated lncRNAs: senescence-associated long noncoding RNAs
- 2014 Regulation of metabolism by long, non-coding RNAs
- 2012 Transgenerational glucose intolerance with Igf2/H19 epigenetic alterations in mouse islet induced by intrauterine hyperglycemia
Antisence Long Non-coding RNAs that are Differentially Regulated with the induction of Cellular Senescence
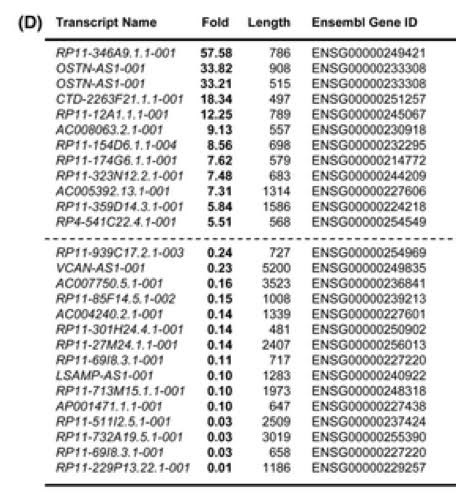
Summary: It is likely that much of the regulation of protein-coding genes and the unique aspects of humans, human aging, and human disease may be due to the “invisible human genome”, which is the large number of non-coding RNAs. This field is exploding, with new RNA sequences being discovered every day. Already, an Antisense strand long noncoding RNA has been discovered at the SIRT1 gene, which increases SIRT1 gene expression by a yet unknown mechanism. One possibility is that this lncRNA prevents or inhibits the functions of miRNAs which increase SIRT! mRNA degradation. Other lncRNAs have been found that regulate cellular senescence, metabolism, etc. via “trans regulatory” or “cis regulatory” methods.
5. Nicotinamide (Nam)
Nicotinamide is a direct inhibitor of both the SIRT enzymes and the PARP enzymes.
The accumulation of excess nicotinamide in cells is probably a major cause of aging. Whereas we typically associate “NAD deficiency” with aging, “Nam excess” may have a similar effect. To no one’s surprise, the levels of the two compounds are inversely related in aging.
Nam plays a role in both aging and in disease. In hypertension and in aging individuals with normal blood pressure, Nam inhibits the methylation-mediated degradation of catecholamines. Thus Nam excess plays a role in hypertension (see references below).
Nicotinamide also has an epigenetic effect. When SIRT1 is inhibited, cells age and cancer oncogenes are re-activated. SIRT1 silences these genes by histone deacetylation of H3K9 and H4K16 residues on the histones of these oncogenes.
A recent article showed that in rats, nicotinamide supplementation during pregnancy causes global DNA hypomethylation in rat fetuses. Nicotinamide has detrimental effects in development, detrimental metabolic effects, and detrimental epigenetic effects when given to young rats. Low dose nicotinamide increased weight gain in developing rats. High dose nicotinamide did not, however. The livers of nicotinamide-fed young rats had more DNA damage (8oxoG), impaired glucose tolerance, and increased insulin resistance. Nicotinamide increased the levels of N-methylnicotinamide in the blood and decreased betaine levels in the blood. This resulted in a global hypomethylation of DNA in the rat genome. Nicotinamide also had “gene-specific effects” on CpG islands within the promoters of the following genes:
- NNMT gene – this was down-regulated
- DNMT genes – these were down-regulated
- Homocysteine metabolism genes – these were down-regulated
- Antioxidant genes and oxidative stress protection genes – these were down-regulated
Since niacin is converted into nicotinamide in human tissues, high dose niacin probably produces all of the above effects. A recent paper called niacin and nicotinic acid “methyl consumers” and strongly suggested that high niacin/nicotinic acid intake is bad.
Excess nicotinamide has also been shown to increase plasma serotonin and histamine levels in humans, due to disrupting the metabolism of these neurotransmitters. This is probably due to the fact that methyl donors and methylation enzymes are needed for serotonin/histamine metabolism. Most importantly, nicotinamide is a direct inhibitor of the Sirtuin enzymes (SIRT1-7) and the PARP enzymes (all 17 of the PARPs).
The molecular mechanism by which Nam works is very straightforward. Nam acts as a direct inhibitor of the SIRT1 enzyme pocket where NAD binds. Thus Nam is a “competitive inhibitor” of NAD and is “bad” when it comes to most cancers, aging, and most diseases.
On the other hand, inhibition of SIRT1 by Nicotinamide may be a “good thing” in the brain, where it may prevent NAD+ depletion and thereby protect neurons against excitotoxicity and neuronal cell death induced by PARP1.
As the cell consumes NAD (by SIRT1-7, PARP1, PARP2, Tankyrases, CD38, CD157, ARTs, and other enzymes), the NAD is consumed, leaving the by-product, Nam. There are two primary ways of “disposing of Nam”. They are methylation/excretion or recycling of Nam into NMN (and subsequently NAD) via the “NAD salvage cycle”.
Here are the problems with both of these methods of reducing Nam levels in the cell. (see #2 and #3 below).
References:
- 2002 Inhibition of Silencing and Accelerated Aging by Nicotinamide, a Putative Negative Regulator of Yeast Sir2 and Human SIRT1
- 2006 Inhibition of SIRT1 Reactivates Silenced Cancer Genes without Loss of Promoter DNA Hypermethylation
- 2009 Nicotinamide Prevents NAD+ Depletion and Protects Neurons Against Excitotoxicity and Cerebral Ischemia: NAD+ Consumption by SIRT1 may Endanger Energetically Compromised Neurons
- Nicotinamide supplementation induces detrimental metabolic and epigenetic changes in developing rats
- 2013 Association of Nicotinamide-N-Methyltransferase Gene rs694539 Variant with Patients with Nonalcoholic Steatohepatitis
- 2013 Excess nicotinamide increases plasma serotonin and histamine levels
- 2013 Excessive nicotinic acid increases methyl consumption and hydrogen peroxide generation in rats
- 2014 Metabolism: Targeting a fat-accumulation gene
Conclusion: It is now clear that high concentrations nicotinamide are harmful to health. HIgh doses of dietary niacin probably produce the same effects, despite the many benefits of high dose niacin. With aging, nicotinamide levels already go up. Adding more nicotinamide is probably not going to “cure” aging. Adding a methyl donor to eliminate nicotinamide (such as betaine) may be a good thing.
6. NAMPT
-NAMPT is the rate-limiting step in the NAD Salvage Cycle, and is regulated in a circadian fashion.
The enzyme that converts Nam back into NMN in the “NAD salvage cycle” is Nicotinamide phosphoribosyl transferase, or NAMPT. Unfortunately, NAMPT is regulated by circadian rhythms and is primarily up-regulated at night. It is inhibited by not sleeping, however. It is also down-regulated by eating and sedentary lifestyle. Fasting and exercise dramatically up-regulate NAMPT. I really do not see how NMN or NR will really change the gene expression of NAMPT. If anything, since NMN is the “product” of the NAMPT enzyme, high levels of NMN may actually have a “feedback inhibition effect” on NAMPT, just like Nictoinamide has a “feedback inhibition effect” on SIRTs and PARPs.
Paradoxically, in a recent study in zebrafish, resveratrol actually DECREASES the expression of NAMPT. This may be via a “feedback inhibition effect”, since SIRT1 “auto regulates” its own gene expession (see references). Another interesting “twist” is that Angiotensin II receptor blockers (ATR type 1 blockers) actually increase NAMPT gene expression. This may be the molecular mechanism behind the longevity effects of ATR1 blocker medications like Telmisartan.
References:
- 2009 Circadian Clock Feedback Cycle Through NAMPT-Mediated NAD+Biosynthesis
- 2008 Glucose Restriction Inhibits Skeletal Myoblast Differentiation by Activating SIRT1 through AMPK-Mediated Regulation of Nampt
- 2009 Circadian Control of the NAD+ Salvage Pathway by CLOCK-SIRT1
- 2012 Modulatory effect of resveratrol onSIRT1, SIRT3, SIRT4, PGC1α andNAMPT gene expression profiles in wild-type adult zebrafish liver
- 2009 Disruption of the Ang II type 1 receptor promotes longevity in mice
- Metabolism: Targeting a fat-accumulation gene
7. NNMT – Methylation of Nicotinamide – Nam)
Methylnicotinamide is a “mitohormetic compound” that regulates longevity
Of all of the unusual aspects of Sirtuins and NAD metabolism, N-methylnicotinamide is probably the hardest one to understand. Recent evidence has shown that for the longevity effects of NAD metabolism to occur in nematodes (C. elegans), NAM must be methylated and then used by the gene, GAD-3, to produce low levels of hydrogen peroxide, thereby acting as a “mitohormetic compound”. This low level of hydrogen peroxide induces mitochondrial biogenesis and is necessary for nematode lifespan extension.
Reference: 2013 Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide
This article came out last year and one of the co-authors is David Sinclair.
However, that is not the entire story. There is much more to the story of the methylation of nicotinamide. Most of this has to do with stopping the toxic effects of nicotinamide from occurring, due to the inhibitory effects of Nam on both the Sirtuin enzymes (SIRT1-7) and the PARPs (1-17).
Over 85% of transmethylation reactions occur in the liver, including the methylation of nicotinamide. With the oral intake of nicotinamide, the liver could either methylate the nicotinamide or convert it to NAD. From many studies, it is clear that the liver preferentially methylates nicotinamide, rather than converting it to NAD. The reason why the liver methylates nicotinamide is that high concentrations of nicotinamide cause cell injury and cell death, since nicotinamide inhibits all 7 of the Sirtuins and also inhibits the PARP enzymes (PARP1,PARP2, etc).
For this reason, with oral intake of Nam, serum levels of N-methylnicotinamide increase. Only 15% of nicotinamide methylation occurs outside of the liver. Nicotinamide methylation by NNMT also requires a methyl donor, such as SAMe or trimethylglycine, which is also called betaine. When nicotinamide is methylated, it is then excreted, thereby reducing Nam levels in the body and preventing SIRT/PARP inhibition.
GWAS studies of polymorphisms in the NNMT gene have revealed an amazing association with Non-alcoholic steatohepatitis (NASH). The NNMT gene SNP, rs694539, is a SNP found in the regulatory portion of NNMT gene. The “GG” genotype protects against NASH, with an OR of 0.58. The “AA” genotype increases the risk of NASH with an OR of 7.3. This suggests that methylation of nicotinamide is an important factor in preventing NASH (see references below). This same polymorphism (rs694539) has been linked to bipolar disorder recently. The association was “female gender-specific” and did not influence male bipolar risk.
A recent novel theory discussing how “too much NNMT” and “too little NNMT” may both play a role in aging and disease has been proposed. This theory suggests that excess dietary nicotinic acid consumption results in molecular/cellular “methyl consumption” and plays a role in disease. This new theory has been published in Nature and is being taken seriously. The main hypothesis is the increase in “methyl consuming compounds” in our diet contributes to metabolic syndrome and many other “man-made diseases”. Another aspect of this new theory is that NNMT is a “fat accumulation gene” (see references below), since the expression of NNMT is unregulated with increased dietary intake of food, especially foods that are rich in niacin and nicotinic acid. Specifically, the expression of the NNMT gene correlates with the percentage of fat in 20 different mouse strains. The main cause of the up regulation of the NNMT gene is overeating/overfeeding. In fact, Kraus and colleagues recently showed that the administration of methylnicotinamide inhibited NNMT and this increased NAD levels and SAM-dependent gene expression.
References:
- 2012 Nicotinamide, NAD(P)(H), and Methyl-Group Homeostasis Evolved and Became a Determinant of Ageing Diseases: Hypotheses and Lessons from Pellagra
- 2013 Excessive nicotinic acid increases methyl consumption and hydrogen peroxide generation in rats 2011 Dietary methyl-consuming compounds and metabolic syndrome
- 2013 Excessive nicotinic acid increases methyl consumption and hydrogen peroxide generation in rats
- 2014 Targeting a fat-accumulation gene
This theory has been proposed because of recent studies which show a paradoxical effect occurs when NNMT is over-expressed. When this occurs, the NNMT over-expression results in diet-induced obesity. This has been shown in humans and in animal models. For instance, the NNMT gene is unregulated in fat cells with obesity and T2DM. When the NNMT gene is “knocked down”, it protects against diet-induced obesity.
Other references for nicotinamide methylation:
- 1973 Nicotinamide methyltransferase and S-adenosylmethionine:5′-methylthioadenosine hydrolase. Control of transfer ribonucleic acid methylation
- http://www.nature.com/nchembio/journal/v9/n11/full/nchembio.1352.html 2013 Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide
- 1994 Nicotinamide methylation in patients with cirrhosis
- 1984 Nicotinamide methylation tissue distribution, developmental and neoplastic changes
- 1984 Nicotinamide methylation and its relation to NAD synthesis in rat liver tissue culture: Biochemical basis fro the physiological activities of 1-methylnicotinamide
- 1965 Inhibition of the methylation of nicotinamide by chlorpromazine
- 1973 Nicotinamide methyltransferase and S-adenosylmethionine:5′-methylthioadenosine hydrolase. Control of transfer ribonucleic acid methylation
- 2012 Toxic effects of nicotinamide methylation on mouse brain striatum neuronal cells and its relation to manganese
- 2013 Association of Nicotinamide-N-Methyltransferase Gene rs694539 Variant with Patients with Nonalcoholic Steatohepatiti
- 2015 Association of nicotinamide-N-methyltransferase mRNA expression in human adipose tissue and the plasma concentration of its product, 1-methylnicotinamide, with insulin resistance2013 Association of nicotinamide-N-methyltransferase (NNMT) gene rs694539 variant with bipolar disorder
- 2014 Metabolism: Targeting a fat-accumulation gene
Here is the recent (2014)article on NNMT knock-down and the protection of diet-induced obesity: Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity
8. PARP-1 and PARP-2
PARPs are the #1 Intracellular “NAD consumers” . PARP-2 is also a direct inhibitor of SIRT1.
- The Poly-ADP-ribose polymerases (PARPs) are a large family of enzymes involved with DNA damage detection, DNA damage repair, and also cellular apoptosis. They are voracious “NAD consumers” and use as much as 100-150 molecules of NAD when activated by one DNA break. As a result, cells can become NAD depleted in the nucleus, where the PARPs reside. They are also voracious “ATP consumers” and use up as much as 100-150 molecules of ATP when activated by one DNA break. As a result, cells can become ATP depleted, which then induces cellular death. Thus PARPs may be an important way of killing a cell by “suicide,” if there is too much DNA damage.
- PARP-1 is not a direct inhibitor of SIRT1, but because it consumes so much more NAD than Sirtuins do, PARP1 inhibitors or PARP-1 “knock down” have been shown to increase SIRT1activity in cells.
- PARP-2, on the other hand, is a direct negative regulator of SIRT1, independent of NAD activity. When PARP-2 activity increases, which it does with aging (by 10-fold), SIRT1 is inhibited, regardless of NAD levels. However, the deletion of the PARP-2 gene results in hepatic cholesterol accumulation and decreased HDL lipoproteins.
References:
- 2011 PARP-1 Inhibition Increases Mitochondrial Metabolism through SIRT1 Activation
- 2013 PARP-2 knockdown protects cardiomyocytes from hypertrophy via activation of SIRT
- 2011 PARP-2 Regulates SIRT1 Expression and Whole-Body Energy Expenditure
- 2014 Pharmacological Inhibition of Poly(ADP-Ribose) Polymerases Improves Fitness and Mitochondrial Function in Skeletal Muscle
- 2011 Interference between PARPs and SIRT1: a novel approach to healthy ageing?
- 3013 PARP-2 regulates cell cycle-related genes through histone deacetylation and methylation independently of poly(ADP-ribosylation
- 2014 MicroRNA-149 Inhibits PARP-2 and Promotes Mitochondrial Biogenesis via SIRT-1/PGC-1α Network in Skeletal Muscle
- 2014 Deletion of PARP-2 induces hepatic cholesterol accumulation and decrease in HDL levels
- · 2014 Antagonistic crosstalk between SIRT1, PARP-1, and -2 in the regulation of chronic inflammation associated with aging and metabolic diseases
Conclusion: There is probably an “antagonistic cross talk” between SIRT1, PARP-1, and PARP-2 due to their mutual demand/need for NAD. With aging, there is a “sterile inflammation” that occurs, often referred to as “inflammaging”. Inflammaging appears to be directly under the control of the NF-kB transcription factor with antagonistic crosstalk between SIRT1, PARP1, and PARP2 signaling pathways. There may be a role for a PARP inhibitor for health. Pharmacological inhibition of PARPs has already been shown to improve skeletal muscle fitness and mitochondrial function in rodent models. In Part 4 of this NAD World series, we will discuss inflammationaging and the roles of NF-kB, NAD and SIRT1 in much further detail
9. CD38
CD38 is probably the #1 Extracellular “NAD consumer” (whereas PARPs are the #1 intracellular consumer)
CD38 is one of several “ectoenzymes” found outside the cell that are “NAD consumers”. (The others are CD157, ART1, ART2, ART3, and ART4). CD38 is a multifunctional membrane-bound extracellular enzyme that plays a key role in immunity, autoimmunity, and calcium signaling. CD38 consumes NAD and makes cyclic-ADP ribose (cADPR). Unfortunately, CD38 is a very inefficient enzyme and consumes as many as 100 NADs for every one cyclic-ADP-ribose that it makes. For this reason, some experts on CD38 feel that the #1 function of CD38 is to regulate cellular NAD levels. A strong argument for this theory is the recent discovery that CD38 is found inside the cell as well, bound to membranes on the inner portion of the cell nucleus. Here it could deplete nuclear NAD. Interestingly, the apple skin-derived flavanoid, apigenin, is a powerful inhibitor of CD38. Treatment of cell cultures with apigenin increased NAD levels in the cells, reduced global acetylation of proteins, and reduced the acetylation of p53 and RelA-p65 subunits of NF-kB.
The classic description of CD38 has not been that of an “intracellular NAD level regulator”, but part of a signaling system involving cyclic ADP-ribose and calcium. Cyclic-ADP-ribose (cADPR) functions as a second messenger in the cell to trigger calcium release from the sarcoplasmic reticulum. CD38 is a very important membrane-bound enzyme found on the surface of many cells, but the highest density of CD38 are on the surface of immune cells such as monocytes/macrophages.
In the brain, CD38 is very important for the secretion of oxytocin from oxytocin-producing cells in the hypothalamus. Oxytocin has traditionally been thought of as the “maternal milk/nurturing hormone”, but in the brain it functions as a neurotransmitter. In the brain, there are many locations for oxytocin type 1 (OTR1) and oxytocin type 2 (OTR2) receptors, located mostly in the telencephalon. Oxytocin appears to be the “Peptide that binds our hearts in love”. It has clearly been linked to maternal love, brotherly love, spousal/opposite sex affection, and community spirit. It may be the peptide that is secreted in response to religious experiences of love for God as well.
References:
- 2009 CD38 as a regulator of cellular NAD: a novel potential pharmacological target for metabolic conditions
- 2002 CD38 expression is an important prognostic marker in chronic lymphocytic leukaemia
- CD38 expressed on human monocytes: A coaccessory molecule in the superantigen-induced proliferation
- 2004 CD38 expression on CD8 T lymphocytes as a marker of residual virus replication in chronically HIV-infected patients receiving antiretroviral therapy
- 2005 CD38 expression levels in chronic lymphocytic leukemia B cells are associated with activation marker expression and differential responses to interferon stimulation
Here is a diagram that illustrates the possible way that CD induces NAD depletion and therefore causes metabolic syndrome:

Ref for diagram: CD38 as a regulator of cellular NAD: a novel potential pharmacological target for metabolic conditions
” Possible mechanism of regulation of SIRT1 and AMPK pathway by CD38 inhibition.
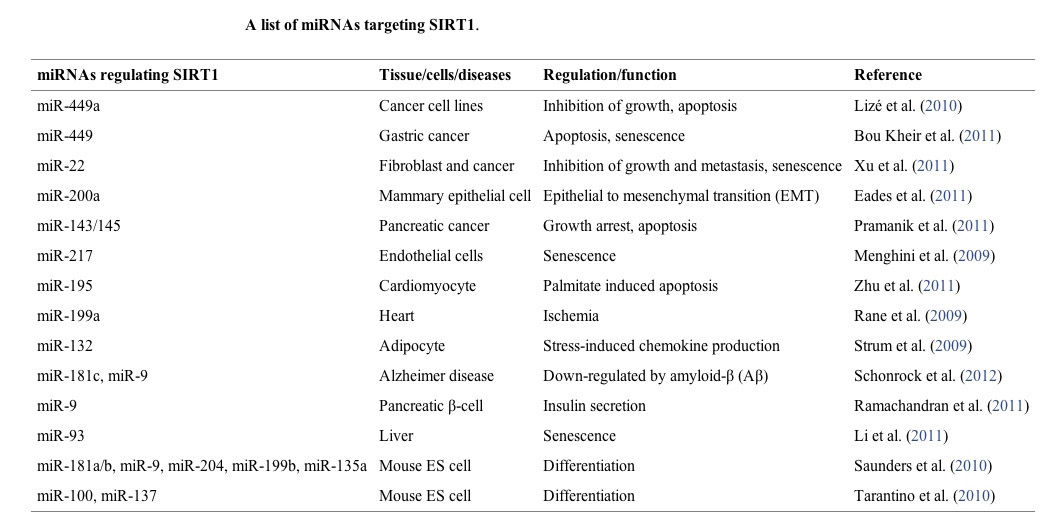
10. MicroRNAs
16 microRNA have been found to regulate SIRT1 expression. These miRNA are mostly negative regulators of SIRT1 expression by their binding to the 3′ UTR of SIRT1 mRNA, increasing SIRT1 mRNA degradation before they can be transcribed. Different microRNA are expressed by different triggers, such as EtOH, p53, and Diabetes type II.
16 different micrRNAs have been found that regulate SIRT1 expression (see table below). Different triggers induce the expression of each miRNA. These microRNA all down-regulate SIRT1 expression. This occurs because microRNA bind to a region in the 3′ untranslated region (3′ UTR) in the SIRT1 mRNA, thereby increasing the degradation of SIRT1 mRNA. Thus microRNA are a “post-transcriptional regulators” of SIRT1 gene expression. This miR-mediated effect is likely the mechanism by which cancer increases the expression of SIRT1 and also the mechanism by which aging decreases the expression of SIRT1.
miR-217 – an miR that is activated by drinking EtOH
Ethanol also activates miR-217 gene expression and is one of the primary mechanisms responsible for alcoholic fatty liver disease and NASH (which is the clinically symptomatic form of alcoholic fatty liver disease). NASH can lead to alcoholic cirrhosis, liver failure, ascites, and death. miR-217 is also the mechanism by which HIV infections shorten lifespan, since tat-activated LTR expression induces miR–217.
miR-34a – an miR that is activated by p53
miR-34a has been the most studied microRNA when it comes to SIRT1 expression. It is involved in pancreatic cancer, colorectal cancer, prostate cancer, brain cancer, liver cancer, normal neural differentiation, liver metabolism, endothelial cell senescence, and endothelial progenitor cell senescence.
Ectopic miR-34a reduces SIRT1 expression. The gene for miR-34a is “turned on” by p53 protein. miR-34a has been shown to induce cancer cell apoptosis in colon cancer cells. miR-34a also promotes endothelial cell senescence in atherosclerosis.
miR-181a – an miR that is increased in diabetes type II
miR-181a binds to a region in the 3′ untranslated region (3′ UTR) in the SIRT1 mRNA, thereby increasing the degradation of SIRT1 mRNA. Thus miR-181a is a “post-transcriptional regulator” of SIRT1 gene expression. Over-expression of miR-181a results in insulin resistance. Studies of the serum (plasma) of diabetics has shown that miR-181a is increased in the serum. Studies of diabetics has also shown that miR-181a is increased in hepatocytes. There is hope that inhibiting miR-181a may be a strategy for treating diabetes type II.
There are many other miRNAs that regulate SIRT1, listed below.
References:
- 2012 MicroRNA Regulation of SIRT1
- 2009 MicroRNA 217 Modulates Endothelial Cell Senescence via Silent Information Regulator 1
- 2008 miR-34a repression of SIRT1 regulates apoptosis
- 2009 Cellular Regulation of SIRT1
- 2012 MiR-217 is involved in Tat-induced HIV-1 long terminal repeat (LTR) transactivation by down-regulation of SIRT
- 2012 MicroRNA-217 Promotes Ethanol-induced Fat Accumulation in Hepatocytes by Down-regulating SIRT1
- 2010 MicroRNA-34a regulation of endothelial senescence
- 2012 Downregulation of miR-181a upregulates sirtuin-1 (SIRT1) and improves hepatic insulin sensitivity
- 2012 MicroRNA Regulation of SIRT1
11. p53
There are two binding sites for p53 on the promoter of SIRT1. They are located at positions 168 and 178 upstream from the TSS. p53 binding to the SIRT1 promoter prevents gene expression of SIRT1, except with starvation. With starvation stress, p53 dissociates from the SIRT1 promoter and FoxO3a can “knock p53 off these two binding sites (168 and 178), thereby removing the repressive effects of p53 from the SIRT1 promoter, thereby resulting in SIRT1 gene expression.
References:
- 2010 SIRT1 and p53, effect on cancer, senescence and beyond
- 2014 SIRT1 phosphorylation by AMP-activated protein kinase regulates p53 acetylation
12. FoxO3a and Eating (especially sugar):
Glucose induced Insulin signaling activates the Insulin/IR/IRS-1/PIP3K/Akt pathway which prevents Foxo3a from migrating into the cell nucleus and activating the SIRT1 gene. Fasting does the opposite. FoxO3a also enters the nucleus with fasting and “bumps” the two p53s off the two binding sites on the SIRT1 promoter, thereby abolishing the repression of SIRT1 by p53.
Reference: 2004 Stress-Dependent Regulation of FOXO Transcription Factors by the SIRT1 Deacetylase
13. E2F1
The cell cycle and apoptosis regulator.
In actively growing cells, E2F1 is the transcription factor which controls cell-cycle fluctuations in SIRT1 levels. E2F1 induces SIRT1 gene expression in response to other factors also, such as cellular oxidative stress from exercise, low dose ETOH, chemo, XRT, and maybe your “oxidative stress water.” If this water would work as a “controllable timeable ROS dose,” this is the molecular mechanism by which it would work. OBTW, SIRT1 also has a negative feedback loop, inhibiting E2F1.
References:
- 2012 SIRT1: new avenues of discovery for disorders of oxidative stress
- 2011 Transcriptional and Nontranscriptional Functions of E2F1 in Response to DNA Damage
- 2014 AMP-activated Protein Kinase α2 and E2F1 Transcription Factor Mediate Doxorubicin-induced Cytotoxicity by Forming a Positive Signal Loop in Mouse Embryonic Fibroblasts and Non-carcinoma Cells
- Targeting cardiovascular disease with novel SIRT1 pathways
- 2014 Sirt1 is a tumor promoter in lung adenocarcinoma
14. HIC1:CtBP co-repressor complex
The SIRT1 promoter has a binding site for a co-repressor complex called “HIC:CtBP”, which decreases SIRT1 gene expression. With CR and fasting or “fasting Mimetics like 2-DG”, the affinity of CtBP for HIC goes down, thereby increasing SIRT1 gene expression several fold. This is one of the primary molecular mechanisms of fasting.
References:
- 2012 Molecular dissection of the interaction between HIC1 and SIRT1
- 2012 Hypermethylation of HIC1 Promoter and Aberrant Expression of HIC1/SIRT1 Might Contribute to the Carcinogenesis of Pancreatic Cancer
- 2013 Identification of p21 (CIP1/WAF1) as a direct target gene of HIC1 (Hypermethylated In Cancer 1)
- 2013 Signification of Hypermethylated in Cancer 1 (HIC1) as Tumor Suppressor Gene in Tumor Progression
15. ChREBP
The carbohydrate response element binding protein.
ChREBP, is up-regulated with the dietary intake of carbohydrates. ChREBP is the “molecular link” between carbohydrate ingestion and high triglycerides in the blood. (i.e. the glucose-induction of triglyceride synthesis.) How does this occur? Well, ChREBP represses the transcription of SIRT1.
ChREBP may play a major role in alcoholism and alcohol-induced fatty liver disease. In a mouse model of binge drinking, ChREBP acetylation was increased dramatically and was recruited to gene promoters in mice. The acetylation of ChREBP was dependent on alcohol metabolism rate. In mice with mutant forms of ChREBP that could not be acetylated, the ChREBP-dependent genes could not be “turned on”. ChREBP silencing in mice that were fed alcohol prevented the increase in triglycerides that normally occurs with binge drinking. In addition, SIRT1 was down-regulated in these mouse models of EtOH binge drinking, due to the direct inhibitory effect of ChREBP on SIRT1 gene expression.
References:
- 2011 CREB and ChREBP oppositely regulate SIRT1 expression in response to energy availability
- 2015 Novel role for ChREBP in the control of ethanol metabolism and susceptibility to binge drinking
- 2015 Novel role for ChREBP in the control of ethanol metabolism and susceptibility to binge drinking
- 2013 Novel insights into ChREBP regulation and function
- 2012 The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans
- 2014 Liquid fructose downregulates Sirt1 expression and activity and impairs the oxidation of fatty acids in rat and human liver cells
- 2012 Glucose sensing by ChREBP/MondoA–Mlx transcription factors
Conclusions: ChREBP is a “glucose sensor”. ChREBP is the direct molecular link between high fructose or high glucose intake and the hepatic formation of triglycerides. ChREBP is also a major inhibitor of SIRT1 gene transcription. More importantly, ChREBP is the “binge drinking gene.”
16. CREB
The cyclic-AMP response element binding protein,
CREB, activates SIRT1 gene expression. This is activated by low glucose levels, which happens with fasting. CREB may also be the transcription factor that explains some of the paradoxical effects of SIRT1 over-expression. In mice that over-express SIRT1, an atherogenic diet does not worsen glucose metabolism. Instead, SIRT1 over-expression protects these mice fed an atherogenic diet from glucose dysregulation (i.e. insulin resistance).
However, in these SIRT1 over-expressed mice on an atherogenic diet, their atherosclerotic lesions actually get much worse than controls. The reason for this is that SIRT1 deacetylates CREB, preventing its cyclic-AMP phosphorylation. Thus SIRT1 inhibits CREB from activating gluconeogenic genes and inhibits CREB from activating hepatic lipid metabolism and excretion. In summary, CREB activates SIRT1 gene expression, and SIRT1 has a negative feedback effect on CREB function. This explains how SIRT1-mediated CREB deacetylation regulates the balance between glucose and lipid metabolism.
ReferenceS:
- 2011 CREB and ChREBP oppositely regulate SIRT1 expression in response to energy availability
- 2011 Proatherogenic Abnormalities of Lipid Metabolism in SirT1 Transgenic Mice Are Mediated through Creb Deacetylation
- 2011 Sirt1 Mediates Neuroprotection from Mutant Huntingtin by Activation of TORC1 and CREB Transcriptional Pathway
- 2011 Sirt1 Mediates Neuroprotection from Mutant Huntingtin by Activation of TORC1 and CREB Transcriptional Pathway
Conclusion: CREB is a “glucose and lipid sensor” with reciprocal interactions with SIRT1. CREB activates SIRT1, which is the opposite of ChREBP. Fasting activates CREB, whereas fasting inhibits ChREBP. Moreover, when there is too much fat, but SIRT1 is over-expressed, CREB cannot prevent atherosclerosis and atherosclerosis worsens.
17. TLX
TLX is one of the “orphan nuclear receptors”.
TLX binds to a TLX-response element in the SIRT1 promoter. TLX is short for “Tailess.”
TLX is a very important transcriptional repressor in the brain, especially in neural stem cells and is vital to normal growth and development. As of now, no endogenous ligand for these ligand-dependent transcription factors has been identified. It is a “druggable” target, however.
So far, only three compounds have been found out of a 20,000 compound high-throughput screen that bind to TLX (ccrp1, ccrp2, and ccrp3). Although many functions for TLX have been discovered, the main cellular function of TLX appears to be keeping neural stem cells in their undifferentiated, proliferative state. TLX regulates the expression of another nuclear receptor, the “retinoic acid receptor” or RAR. Thus TLX is an important receptor. TLX is an oncogene-induced senescence suppressor inside and outside of the brain. It has been shown to be effective in the prosate.
TLX co-regulates the cyclin-D kinase inhibitor, CDKN1A (aka p21WAF/CIP1) with SIRT. Very little is known about this orphan nuclear receptor other than the fact that it activates SIRT1 gene expression.
References:
- 2015 Orphan nuclear receptor TLX functions as a potent suppressor of oncogene-induced senescence in prostate cancer via its transcriptional co-regulation of the CDKN1A (p21WAF1/CIP1) and SIRT1 genes
- 2015 TLX: A master regulator for neural stem cell maintenance and neurogenesis
- 2013 Insulin induces neurite outgrowth via SIRT1 in SH-SY5Y cells
- 2014 SIRT1 is required for oncogenic transformation of neural stem cells and for the survival of “cancer cells with neural stemness” in a p53-dependent manner
- 2008 Pathological aggression in “fierce” mice corrected by human nuclear receptor 2E1
- 2014 The Human Orphan Nuclear Receptor Tailless (TLX, NR2E1) Is Druggable
- 2004 Expression and function of orphan nuclear receptor TLX in adult neural stem cells
18. C/EBP-alpha and beta
C/EBP-alpha and C/EBP-beta – the two hepatic CCAAT/enhancer binding proteins with opposite effects on SIRT1/
In the liver, there are two opposing transcription factors that play a major role in liver biology, glucose metabolism, and fat metabolism: CCAAT/enhancer binding proteins alpha and beta. These two transcription factors are also expressed elsewhere, outside of the liver, but the general role of the two appear to be similar – they have opposing roles on gene expression.
- The CCAAT/enhancer-binding protein alpha (C/EBP-alpha) is a transcription factor that represses many genes and activates many genes. The most important 3 are SIRT1, p53, and PGC-1a.
- There is also a C/EBP-beta that has the opposite effects on these genes.
For instance C/EBP-alpha represses the hTERT gene, thereby preventing cancer induction of telomerase. C/EBP-beta, on the other hand increases the expression of the hTERT gene, thereby increasing telomerase enzymes.
C/EBP-alpha activates SIRT1 gene expression via binding to the promoter region of SIRT1. The opposite is true about C/EBP-beta. It represses SIRT1 gene expression.
In old age, SIRT1 cannot be up-regulated very well due to the repressor effects of the C/EBP-beta/HDAC1 complex, which both work together to suppress SIRT1 gene expression.
Interestingly, pomegranate seed oil has 3 ingredients in it (xanthigen, fucoxanthin, and punicic acid) that appear to down regulate C/EBP-beta and thus prevent fat accumulation. Xanthigen up regulates SIRT1 and AMPK signaling in differentiated fat cells also.
References:
- 2012 CEBP factors regulate telomerase reverse transcriptase promoter activity in whey acidic protein-T mice during mammary carcinogenesis
- 2014 Terminating hepatocyte proliferation during liver regeneration: The roles of two members of the same family (CCAAT-enhancer-binding protein alpha and beta) with opposing actions
- 2012 Transcription factor CCAAT/enhancer-binding protein alpha and critical circadian clock downstream target gene PER2 are highly deregulated in diffuse large B-cell lymphoma 2012 Xanthigen Suppresses Preadipocyte Differentiation and Adipogenesis through Down-regulation of PPARγ and C/EBPs and Modulation of SIRT-1, AMPK, and FoxO Pathways
- 2013 Transcriptional and Translational Regulation of C/EBPβ-HDAC1 Protein Complexes Controls Different Levels of p53, SIRT1, and PGC1α Proteins at the Early and Late Stages of Liver Cancer
- 2011 The reduction of SIRT1 in livers of old mice leads to impaired body homeostasis and to inhibition of liver proliferation†
Conclusion: The two CCAAT/enhancer-binding proteins alpha and beta have important opposing effects on tissue regeneration, glucose metabolism and fat metabolism. In general, C/EBP-alpha prevents fat accumulation and promotes liver regeneration, whereas C/EBP-beta has the opposite effect. C/EBP-alpha increases SIRT1 gene expression, whereas C/EBP-beta and HDAC-1 combine to repress the promoter of SIRT1.
19. BRCA1
BRCA1 increases the expression of NAMPT, PARP1 and SIRT1, whereas BRCA1 mutation, promoter methylation, or knockdown decreases NAMPT, PARP1, and SIRT1 gene expression, but paradoxically increases NAD levels, which then increase SIRT1 activity (but not SIRT1 gene expression). Thus BRCA1 may be a “balancer” between SIRT1 gene expression and SIRT1 protein activity
Two decades ago, the first breast cancer susceptibility gene was discovered and called “BRCA1”. Now we know the BRCA1 protein (which is involved in double stranded DNA repair), is mutated in hereditary forms of breast and ovarian cancer. Inherited BRCA1 mutations can lead to cancers of the breast, ovary, and many other organs. For breast cancer, the risk due to the mutation is increased to 56-80%. For ovarian cancer, the risk is increased to 15-60%. BRCA1 regulates the expression of 7% of the mRNA in cancer cells. This is probably why it is so important and should therefore be called a “transcriptional regulator”.
Although a lot of research was being done about BRCA1 and cancer, no one had linked BRCA1 to Sirtuins or NAD until very recently. Recently, the BRCA1 protein was found to control NAMPT-mediated NAD synthesis, which was a surprise. Another surprise was that NAD levels could have a “feedback activation” of BRCA1 expression. Moreover, it has been shown that BRCA1 is a positive regulator of PARP1 levels and NAD-dependent PARP1 activity. Most recently, BRCA1 was found to also positively regulate SIRT1 expresión. BRCA1 binds to the SIRT1 promoter and activates SIRT1 gene expression. SIRT1 then inhibits the expression of Survin by deacetylating the H3 histones of the Survivin gene. BRCA1 also inhibits Survivin. Without SIRT1 or BRCA1, Survivin levels increase and Cancer develops.
BRCA1 may also play a role in preventing hypertension. One of the major pathways in spontaneous hypertension is the Angiotensin II pathway and the activation of the ATR1 and ATR2 receptor by Angiotensin II. ATR1 receptor activation induces NAD(P)H oxidase-induced free radical formation and the vasoconstriction of blood vessels due to reduced NO production. A recent article showed that BRCA1 limits Angiotensin II ATR1-mediated redox signaling, thereby improving vascular reactivity and reduces blood pressure in spontaneously hypertensive mice.
References:
- 2015 Linking BRCA1 to NAD World
- 2014 A novel crosstalk between BRCA1 and sirtuin 1 in ovarian cancer
- 2012 497 BRCA1 Limits AT1-Mediated Redox Signaling, Improves Vascular Reactivity and Reduces Blood Pressure in Spontaneously Hypertensive Rats
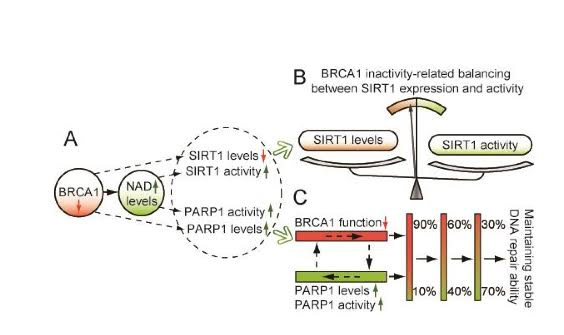 Illustration Reference: 2015 Linking BRCA1 to NAD World “Figure 1. Proposed model of crosstalk among BRCA1, SIRT1 and PARP1. A, BRCA1 inactivation may regulate SIRT1 and PARP1 levels, and induce an increase in NAD-mediated SIRT1 and PARP1 activity. B, the model shows a significant effect of BRCA1 in the maintenance of SIRT1-related biological processes. C, a proposed model to maintain stable BRCA1 and PARP1-related DNA repair ability.”
Illustration Reference: 2015 Linking BRCA1 to NAD World “Figure 1. Proposed model of crosstalk among BRCA1, SIRT1 and PARP1. A, BRCA1 inactivation may regulate SIRT1 and PARP1 levels, and induce an increase in NAD-mediated SIRT1 and PARP1 activity. B, the model shows a significant effect of BRCA1 in the maintenance of SIRT1-related biological processes. C, a proposed model to maintain stable BRCA1 and PARP1-related DNA repair ability.”
20. SNPs in the SIRT1 promoter
Evolution and the “Feast or Famine” SNP in the SIRT1 promoter region in Northern India. rs12778366 is a single nucleotide polymorphism (SNP) found 1.46 kb upstream from the TSS in the SIRT1 promoter that predisposes N Indians to type II diabetes. > 80% of N. Indians have this SNP and have a 6-9 fold higher risk of T2DM. This SNP was probably “selected by famines” that often occurred in N India for 1,000s of years. Now it is a liability, since famines no longer occur
21. EGR1
Mechanical stretching of muscles:
The SIRT1 gene is upregulated by the stretching of muscle fibers via the transcription factor “Early Growth Response Factor 1”, or EGR1. Mechanical stretching of muscles increases mRNA for SIRT1 by 2.2 fold and SIRT1 protein by 100%! This is why stretching before and after exercise is so important, since SIRT1 expression causes FoxO3a deacetylation (and thereby induction of the mitochondrial SOD gene) as well as Nrf2 Deacetylation (and thereby induction of many antioxidant genes).
22. DBC1 (aka CCAR2)
“Deleted in Breast Cancer 1” is probably a bad name for this protein, since it may not even be deleted in most breast cancers.
For this reason, the new name for DBC1 is “Cell cycle activator and apoptosis regulator 2”, or CCAR2. DBC1 directly interacts with SIRT1 by forming a stable complex with DBC1, thereby preventing the activity of SIRT1 in vitro and in vivo. Interestingly, SIRT1 and DBC1 protein levels are higher in breast cancer tissues, compared to age-matched controls, but not at the transcriptional level. At the transcriptional level, there does not appear to be up regulation of SIRT1 and DBC1. This up regulation of SIRT1 and DBC1 is therefore at the “postranscriptional level” (i.e. via microRNA or microRNA sinks). In gastric adenocarcinoma, overexpression of SIRT1 and DBC1 are actually associated with a better prognosis.
Reference:
- 2013 hMOF Acetylation of DBC1/CCAR2 Prevents Binding and Inhibition of SirT1
- 2008 DBC1 is a negative regulator of SIRT1
- 2008 Negative regulation of the deacetylase SIRT1 by DBC1
- 2010 Balance between SIRT1 and DBC1 expression is lost in breast cancer
- 2009 Interactions between DBC1 and SIRT1 are deregulated in breast cancer cells
- 2012 Expression of SIRT1 and DBC1 in Gastric Adenocarcinoma
- 2008 DBC1 is a negative regulator of SIRT1
23. c-Myc
There is now evidence that the SIRT1 gene expression is “downstream” from the oncogene, c-Myc. In cancer, there is a “positive feedback loop” that occurs, which results in contributes to the development of cancer. Specifically, c-Myc activates SIRT1, which in turn promotes SIRT1 function. Likewise, SIRT1 promotes c-Myc function. Here is the article on this. This “mutual positive feedback” between SIRT1 and c-Myc is why there is so much confusion as to whether SIRT1 is a tumor suppressor or a tumor enhancer.
Reference: 2011 The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop
“Silent information regulator 1 (SIRT1) represents an NAD+-dependent deacetylase that inhibits proapoptotic factors including p53. Here we determined whether SIRT1 is downstream of the prototypic c-MYConcogene, which is activated in the majority of tumors. Elevated expression of c-MYC in human colorectal cancer correlated with increased SIRT1 protein levels. Activation of a conditional c-MYC allele induced increased levels of SIRT1 protein, NAD+, and nicotinamide-phosphoribosyltransferase (NAMPT) mRNA in several cell types. This increase in SIRT1 required the induction of the NAMPT gene by c-MYC. NAMPT is the rate-limiting enzyme of the NAD+ salvage pathway and enhances SIRT1 activity by increasing the amount of NAD+. c-MYC also contributed to SIRT1 activation by sequestering the SIRT1 inhibitor deleted in breast cancer 1 (DBC1) from the SIRT1 protein. In primary human fibroblasts previously immortalized by introduction of c-MYC, down-regulation of SIRT1 induced senescence and apoptosis. In various cell lines inactivation of SIRT1 by RNA interference, chemical inhibitors, or ectopic DBC1 enhanced c-MYC-induced apoptosis. Furthermore, SIRT1 directly bound to and deacetylated c-MYC. Enforced SIRT1 expression increased and depletion/inhibition of SIRT1 reduced c-MYC stability. Depletion/inhibition of SIRT1 correlated with reduced lysine 63-linked polyubiquitination of c-Myc, which presumably destabilizes c-MYC by supporting degradative lysine 48-linked polyubiquitination. Moreover, SIRT1 enhanced the transcriptional activity of c-MYC. Taken together, these results show that c-MYC activates SIRT1, which in turn promotes c-MYC function. Furthermore, SIRT1 suppressed cellular senescence in cells with deregulated c-MYC expression and also inhibited c-MYC–induced apoptosis. Constitutive activation of this positive feedback loop may contribute to the development and maintenance of tumors in the context of deregulated c-MYC.
24. AROS
Active Regulator of SIRT1
AROS is an endogenous activator of SIRT1. AROS may bind to the site where resveratrol and other STAC activators bind to SIRT1, but this is still unclear. What is clear is that AROS increases SIRT1 activity and works with SIRT1 to suppress p53 activity. Specifically AROS works with SIRT1 to deacetylate p53, thereby reducing p53-mediated transcriptional activity (gene expression of genes dependent on p53). Thus SIRT1 and AROS are negative feedback regulators of p53.
Reference: 2007 Active Regulator of SIRT1 Cooperates with SIRT1 and Facilitates Suppression of p53 Activity
25. HuR
The Hu protein called HuR is an RNA binding protein that stabilizes the SIRT1 mRNA, preventing its degradation
Many RNA binding proteins have been recently discovered that degrade or stabilize messenger RNA (mRNA). This includes the mRNA degrading RNA-binding proteins, AUF1, BRF1, TTP, and KSRP. The mRNA-stabilizing RNA binding proteins include the elav/Hu proteins, of which HuR is one. HuR is probably the most well-known RNA-binding protein that reduces mRNA degradation. It binds to the SIRT1 mRNA in the cytoplasm to prevent the SIRT1 mRNA from being degraded. Whereas the 16 different miRNA that bind to SIRT1 mRNA all promote its degradation, HuR binds to the same 3′ UTR region on the SIRT1 mRNA, thereby preventing the miRNA-mediated degradation of the SIRT1 mRNA. The net result of HuR is that there is more SIRT1 protein as a result of the same level of SIRT1 gene transcription.
Reference: 2007 Phosphorylation of HuR by Chk2 Regulates SIRT1 Expression
26. JNK2
The c-Jun Kinase, JNK2 phosphorylates the SIRT1 protein, thereby stabilizes the SIRT1 protein
The SIRT1 protein has several phosphorylation sites on Serine amino acid side chains. Ser27 is one of these sites that gets phosphorylated indirectly by JNK2 activation. When the Ser27 site on SIRT1 is phosphorylated, the SIRT1 protein becomes much more resistant to proteasome-mediated degradation. Thus it increases the half life of the SIRT1 protein from < 2 hrs to > 9 hours. This is a very important part of maintaining SIRT1 protein levels within the cell.
Reference: 2008 JNK2-dependent regulation of SIRT1 protein stability
27. Resveratrol, SRT1720, SRT2104, EX527
Resveratrol is a natural STAC activator. Synthetic STAC activators such as SRT1720 have also been synthesize
The initial excitement about Sirtuins was primarily directed towards the natural compound, reseveratrol, found in red grape skins, Japanese knotweed, and many other plants. Resveratrol and other STAC activators only activate SIRT1 and not SIRT2-7 or the PARP enzymes. For this reason, resveratrol may hold certain advantages over NAD therapy or NAD precursor therapy such as NR or NMN. The STAC-activating site on SIRT1 is near amino acid E320. This site is not present on the other 6 isoforms of mammalian SIRT (SIRT2-7).
References:
- Sirtuin-activating compound
- SIRT1 Activators: The Evidence STACks up
- Evidence for a common mechanism of SIRT1 regulation by allosteric activators
- New findings on mechanism of activation of sirtuins may vindicate Sirtris founders
28. Lamin A
Lamin A is part of the nuclear cytoskeleton and may bind to the C-terminus of SIRT1. Resveratrol may activate SIRT1 in a Lamin A-dependent manner.
There is a link between accelerated aging and the Laminopathies. The most well-known laminopathies is Hutchinson-Gilford Progeria Syndrome, or HGPS. In HGPS, a mutation in the Lamin A gene produces a mutant protein called “progerin”, which results in a breakdown of the cytoskeleton of the nuclear matrix. This results in premature aging and usually death due to an MI or stroke during teenage years. Recently, Gosh and colleagues from China have linked Lamin A and SIRT1. According to their work, the C-terminal tail of SIRT1 binds to Lamin-A. Thus lamina A may serve as a “SIRT1 anchor” in the cell nucleus.
Gosh and colleagues also showed that reseveratrol activates SIRT1 in a Lamin A-dependent manner. Specifically, they showed that resveratrol increased the binding of SIRT1 to Lamin A and also down-regulated FoxO3 acetylation (SIRT1 is a known FoxO3 deacetylator). Not all experts agree with this mechanism of action. It has not been verified by a 2nd research laboratory. However, this is very intriguing that reseveratrol may be a compound that could help treat HGPS.
Reference: 2013 Resveratrol activates SIRT1 in a Lamin A-dependent manner
Aging
Aging lowers SIRT1 activity by multiple mechanisms. The primary mechanism was once thought to be due to decreased gene expression of the SIRT1 gene, but this may not be true in the majority of cases. Instead, age-induced reduction in SIRT1 activity is probably due to declining levels of NAD, increasing levels of Nicotinamide, and increasing levels of DBC1.
Reference: 2011 Age Related Changes in NAD+ Metabolism Oxidative Stress and Sirt1 Activity in Wistar Rats
Coming” Part 4 of the NAD world (comment by Vince Giuliano)
| This has been another Chapter in the extended NAD story. But, as usual there is much more to tell. Jim Watson will explain some of that in the forthcoming NAD World Part 4 blog entry, For one matter, there will be a substantial discussion there of the relationship of SIRT1 to inflammation, Also, what might be of prime importance for health and longevity may not be the actual concentration of NAD+ in cells or cell nucli, but rather the NAD+/NAD ratio which may not be affected by NAD precursor supplements and rather driven by other matters such as expression of the NQ01 gene. The forthcoming blog entry will also look into the much-discussed Warburg effect, which appears to be driven by far more than scarcity of NAD+. Jim Watson will discuss factors that can possibly drive the tendency to Warburg-like metabolism with aging, including the expression of three long non-coding RNAs that promote it via three different mechanisms. Also, Jim will touch upon some additional NAD-related intervention strategies that go beyond those normally discussed in the longevity literature, including possible activation of non-coding RNAs. |

Hi,
Sorry, I am the dumbest guy in the room here but I was just wondering, regarding parts of your post in 5. Nicotinamide (Nam)
here:
The livers of nicotinamide-fed young rats had more DNA damage (8oxoG), impaired glucose tolerance, and increased insulin resistance. Nicotinamide increased the levels of N-methylnicotinamide in the blood and decreased betaine levels in the blood. This resulted in a global hypomethylation of DNA in the rat genome
here :
The accumulation of excess nicotinamide in cells is probably a major cause of aging. Whereas we typically associate “NAD deficiency” with aging, “Nam excess” may have a similar effect. To no one’s surprise, the levels of the two compounds are inversely related in aging.
AND….especially here:
Thus Nam is a “competitive inhibitor” of NAD and is “bad” when it comes to most cancers, aging, and most diseases.
Are you saying that NAD+ could be very bad for someone to take? Are you suggesting that it could increase the likelihood of someone developing cancer ( if they were predisposed, etc)? Lastly, any thoughts on how it might affect the immune system? I have sarcoidosis…thought it might help, but after reading this ( with limited understanding), I’m not so sure I should be taking it.
Thanks!
nwetmore:
The negative comments you refer to are with respect to nicotinamide (Nam), not to NAD+. We are not here saying NAD+ could be bad for someone to take. I can understood you getting lost in the alphabet soup. Distinctions between the various NAD-related entities are in the Part 1 blog entry at http://www.anti-agingfirewalls.com/2014/11/11/nad-an-emerging-framework-for-life-extension-part-1-the-nad-world-2/
Vince
@nwetmore – nicotinamide (NAM) is not NAD+. See blog entries part 1 and 2 for clarity on all of the players involved.
@Jim and/or Vincent: Any further comments on the mention that Resveratrol turns off NrF2? This is quite concerning — previous blogs on NrF2 activation listed resveratrol as turning ON NrF2 …http://www.anti-agingfirewalls.com/2012/02/06/the-pivotal-role-of-nrf2-part-2-%e2%80%93-foods-phyto-substances-and-other-substances-that-turn-on-nrf2/
Pingback: 30 Major Factors that Control SIRT1 Expression, SIRT1 Activity, and SIRT1-mediated Aging | Slow and Ultimately Reverse Aging and Get More of the Good Life!
Hello,
Im wondering:
1/ you explain that nicotinamide inhibit SIRT1, but I dont really see evidence for that, on longecity forum we even showed the opposite. You also speak about “excessive nicotinamide” what amount do you think it can represent ? Chromadex showed low dose NR (100mg) worked well to enhance NAD+ within the cells. Thus, if we consume MSM as methyl supplementation, we shouldnt have any problem isnt ?
2/ You also explain that resveratrol only increase SIRT1 expression, and not others sirts. You present it as a good thing. I dont understand because for me best would be increase ALL sirtuins as explained in a previous post you did on your blog.
3/ as explained in this blog: http://www.pterostilbene.com , pterostilbene seems better in all point than resveratrol especially due to its high oil soluble profile and better to activate SIRT1. What do you think about it ?
thanks a lot !
Recently I read an article, in which the relationship of SIRT1 and obesity was discussed. So when talk about the relationship between SIRT1 and aging, I think the three may be connected to each other, as many aged people are kind of fatter than the younger them.
Researches related materials are available at http://www.bocsci.com/
Pingback: The top 21Benefits of Meditation | AGINGSCIENCES™ – Anti-Aging Firewalls™
Pingback: Part 5 of the NAD world: the conflicting roles of NAMPT – inflammation or rescue? Also Part 1 on a new series on inflammation - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™