
By Jim Watson with editorial and minor contributions by Vince Giuliano
Much has been learned over the years about Alzheimer’s Disease (AD) and there are several sides to the unfolding story which considered together are making more and more sense. This blog entry focuses on the role of amyloid beta in the brain, a major factor universally acknowledged to be of importance – although in the absence of agreement as to exactly what that importance is. We advance the hypothesis that a major factor in the progression of the disease is failure of clearance of soluble amyloid beta in the brain. Further, we believe that this is due to lack of adequate binding of APOE species to the LDL receptors. The issue can be some combination of APOE gene polymorphisms which are well known to create AD susceptibly and age-related insufficient production in the brain of the LDL receptor related protein (LRP).
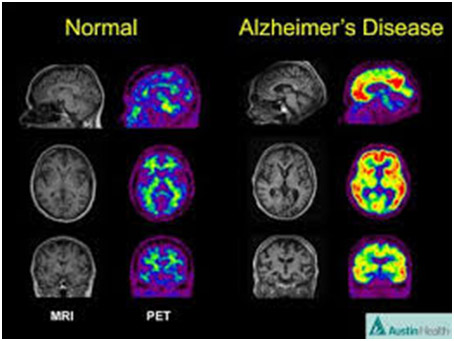
We also discuss several related aspects of the AD story including why presence of beta amyloid is only a possible but not a definitive indicator of AD, details related to how beta amloid can or cannot be created via secretases, the beta-amyloid clearance role of the LDL receptor, age-related inflammaging, the role of the RAGE receptor and AB neurotoxicity, why AB-42 is much more dangerous than AB-40, roles of microglia, how beta-amloid kills neurons and when it does not do that, the roles of APOE and its polymorphisms, and two new high accuracy tests for predicting whether AD is likely to develop..
This blog focuses on these selected aspects of the story. Another compatible part of the story was told in earlier blog entries relating to the role of microglial activation. See Key roles of glia and microglia in age-related neurodegenerative diseases(November 2011), Alzheimer’s Disease Update (March, 2011), and New views of Alzheimer’s disease and new approaches to treating it (February, 2010). Other important parts of the story will be in forthcoming blog entries, including disussion of tau tangles and possible new preventative and therapeutic approaches.
Setting the Context
First, we recapitulate some background on what beta-amyloid is and is is typically involved in cases of AD. The hypothysis that extra-cellular beta amyloid deposits in the brain is usually a key early indicator of AD goes back to the early 90s and has now become quite mainline. The 2002 document The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeuticsrelated “It has been more than 10 years since it was first proposed that the neurodegeneration in Alzheimer’s disease (AD) may be caused by deposition of amyloid beta-peptide (Abeta) in plaques in brain tissue. According to the amyloid hypothesis, accumulation of Abeta in the brain is the primary influence driving AD pathogenesis. The rest of the disease process, including formation of neurofibrillary tangles containing tau protein, is proposed to result from an imbalance between Abeta production and Abeta clearance.” This is still a good formulation of the hypothysis. Problems are, of course, 1. Some people develop high levels of beta-amloid but never progress to showing dementia or AD, and 2. Some people develop dementia thought to be AD but do not have high levels of brain beta-amyloid. In any case for our purposes, if amloid-beta is involved in AD, it is responsible for early stages of the pathology – ones leading to the tau tangles characteristic of later stages.
As time progressed a more nuanced view of the amloid hypothysis has emerged such as laid out in the 2009 publication Reassessing the Amyloid Cascade Hypothesis of Alzheimer’s Disease: “Since its inception, the amyloid cascade hypothesis has dominated the field of Alzheimer’s disease (AD) research and has provided the intellectual framework for therapeutic intervention. Although the details of the hypothesis continue to evolve, its core principle has remained essentially unaltered. It posits that the amyloid-β peptides, derived from amyloid precursor protein (APP), are the root cause of AD. Substantial genetic and biochemical data support this view, and yet a number of findings also run contrary to its tenets. The presence of familial AD mutations in APP and presenilins, demonstration of Aβ toxicity, and studies in mouse models of AD all support the hypothesis, whereas the presence of Aβ plaques in normal individuals, the uncertain nature of the pathogenic Aβ species, and repeated disappointments with Aβ-centered therapeutic trials are inconsistent with the hypothesis. The current state of knowledge does not prove nor disprove the amyloid hypothesis, but rather points to the need for its reassessment. A view that Aβ is one of the factors, as opposed to the factor, that causes AD is more consistent with the present knowledge, and is more likely to promote comprehensive and effective therapeutic strategies.”
SOME KEY PIECES OF THE BETA AMYLOID JIGSAW PUZZLE
As I see it, all the pieces of the Alzheimer’s puzzle are finally starting to fit together. I will try to describe key piecea of the puzzle using some easy-to-understand analogies. Each item below is a piece of the interlocking puzzle. Each piece has at least 4 sides to it as follows:
1. Dispelling the Myth about “Amyloid negative Alzheimer’s disease”
Although the correlation between Amyloid beta accumulation and Dementia is far from perfect, there is no question that Amyloid beta accumulates in over 95% of patients who develop age-related dementia. Today we know that many other distinctly different causes of dementia exist that can present with the exact same signs and symptoms of AD.
As a result, today it is believed that cases of dementia where no Amyloid beta is present on autopsy, one of the following causes of the dementia explain the “Amyloid beta Negative dementia”:
- Vascular dementia
- Lewy body disease
- Chronic traumatic encephalopathy (CTE)
- Frontotemporal dementia
- Parkinson’s disease with dementia (15% of PD)
- Amyotrophic lateral sclerosis (ALS) with dementia
The Misinterpreted Nun Study
Digging a little deeper into the inconclusive relationship of beta amyloid to AD, we go on to dispell the myth of “Amyloid negative Alzheimer’s disease” and provide an explanation for for the Nuns who had Amyloid beta on autopsy, but had no Alzheimer’s disease. (Aka. A slice of the Venn Diagram of Dementia). Our eventual purpose of this blog entry is to lay out what we strongly suspect may be happening.
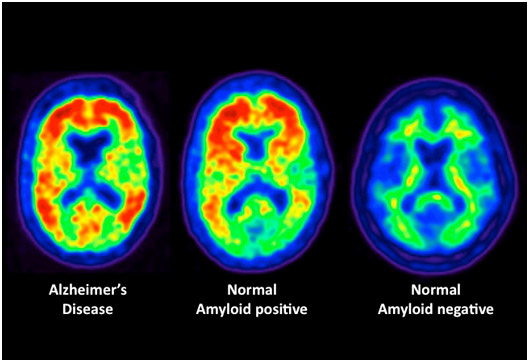
There is a famous “Nun Study” reporting on autopsies done on a series of Catholic Nuns who died. Some had Alzheimer’s disease but many died with no dementia. Although most of the autopsies of Nuns with dementia showed Amyloid beta, some did not. What was even more surprising was that some Nuns with no dementia had amyloid beta accumulation. This study has been often quoted by those who do not believe that amyloid beta causes Alzheimer’s disease. In many ways, this is analogous to the debate of cholesterol and arteriosclerosis. Today most experts do not believe that arteriosclerosis is a “cholesterol disease”. Instead, it is a “cholesterol transport disease” (aka a lipoprotein clearance disease). Likewise, most experts believe that Alzheimer’s is NOT an Amyloid beta disease but a “Amyloid beta clearance disease.” As discussed below, AD may be consequential to possible problems with lipoprotein E and/or the LDL receptor related protein that it binds to for clearing amyloid beta.
Just like the presence of cholesterol does not mean you have arteriosclerosis, the presence of amyloid beta does not mean you have Alzheimer’s disease. This is a key point that most everyone misunderstands about these two diseases. What is even more surprising is how the two “puzzles” (arteriosclerosis and Alzheimer’s) have a common puzzle piece – the LDL receptor! “The Low-Density Lipoprotein (LDL) Receptor is a mosaic protein of 839 amino acids (after removal of 21-amino acid signal peptides)[1] that mediates the endocytosisof cholesterol-rich LDL. It is a cell-surface receptor that recognizes the apoprotein B100, which is embedded in the outer phospholipid layer of LDL particles. The receptor also recognizes the apoE protein found in chylomicron remnants and VLDL remnants (IDL). In humans, the LDL receptor protein is encoded by the LDLR gene.[2][3] It belongs to the Low density lipoprotein receptor gene family[4](ref). ” The LDL receptor on the surface of macrophages in the blood vessel wall and the LDL receptor related protein on the Brain’s microglial cells are almost exactly the same and when these receptors are absent (or decline with aging), there is a 100% incidence of arteriosclerosis and Alzheimer’s disease. Thus many have suggested that we call arteriosclerosis and Alzheimer’s disease both by the same name – “LDL Receptor Disease“. However this is a little bit of an oversimplification, as you will see below.
A person with clinical dementia may not have Alzheimer’s disease, but may have one of the other causes of dementia. This explains “amyloid beta negative Dementia” seen in the famous “Nun study”.
The other part of the “Nun Study”Is also explainable – i.e. the Nuns with no dementia who had Amyloid beta accumulation. This I believe is due to the body’s built in cellular stress coping mechanisms controlled by “gene switches” — transcription factors. The Transcription factors that prevented these Nuns from developing Alzheimer’s disease are named FoxO3a, FoxO1, Nrf2, COREST, PGC-1a, and many other hard-to-understand names. These “amyloid beta positive Nuns” had no dementia because they had developed strong cellular stress resistance. Their neurons were resistant to cell death. This effect is an aspect of hormesis, a non-linear response to stress often discussed in this blog. “. Others like to call it “ischemic preconditioning”. Others explain this phenomena by the molecular mechanisms involved with caloric restriction. (T hey are all different views of the same cellular stress resistance system). Regardless of the terminology used to describe cellular stress resistance, there is no debate that you can reduce cell death by making your brain cells resistant to cell stress.
So, amyloid beta is a “cellular stressor” that can either kill your neurons (and you will develop dementia) or you can become resistant to Amyloid beta toxicity on a cellular level. With aging, everyone develops multiple types of amyloidosis. After age 110, the transthyretin amyloid protein builds up. Are you going to die of “amyloid stress” or are you going to become resistant to”amyloid stress”. This is the question that Hamlet asked himself In the famous Shakesphere line..”to be or not to be.” I would Rephrase this: “to die of amyloid stress or become resistant to Amyloid stress….this is the question. ”
We have discussed hormesis and cellular stress resistance in a number of blog entries. See for example this PowerPoint presentation on it, and the blog entries Multifactorial Hormesis – the theory and practice of maintaining health and longevity, and PRECONDITIONING – Adaptive Response In Biology And Medicine – report on the 2014 annual meeting of the International Dose Response Society.
If you grow old enough, there will be Amyloid beta fibrils present on your autopsy after death. However, your neurons do NOT need to be dead when you die. This explains the “dementia-free, Abeta positive Nuns”.
To avoid mislabeling the cause of dementia, the confirmatory diagnosis of Alzheimer’s disease requires an autopsy that shows histologic evidence of Amyloid beta presenting 8 nm aggregates. This is typically referred to as “amyloid Beta fibrils”. If this is not present, then another cause for the dementia is entertained.
2. Key role of the LDL receptor protein in beta amyloid clearance
We asserted above that AD is involved with inadequate clearance of soluble amyloid beta in the brain. LRP binds to three ligands (ApoE, alpha2 macroglobin, and KPI), two of which have been shown to bind to both AB-40 and AB-42. Both ApoE and alpha2 macroglobulin (a2M) bind to Abeta. Then the ApoE/ABeta or a2M/Abeta binds to the LRP and is endocytosed. This has been known for 14 years, which is somewhat shocking! See the 2000 article Modulation of amyloid β-protein clearance and Alzheimer’s disease susceptibility by the LDL receptor–related protein pathway. “Susceptibility to Alzheimer’s disease (AD) is governed by multiple genetic factors. Remarkably, the LDL receptor–related protein (LRP) and its ligands, apoE and α2M, are all genetically associated with AD. In this study, we provide evidence for the involvement of the LRP pathway in amyloid deposition through sequestration and removal of soluble amyloid β-protein (Aβ). We demonstrate in vitro that LRP mediates the clearance of both Aβ40 and Aβ42 through a bona fide receptor-mediated uptake mechanism. In vivo, reduced LRP expression is associated with LRP genotypes and is correlated with enhanced soluble Aβ levels and amyloid deposition. Although LRP has been proposed to be a clearance pathway for Aβ, this work provides the first in vivo evidence that the LRP pathway may modulate Aβ deposition and AD susceptibility by regulating the removal of soluble Aβ.” –
“Discussion: The genetic associations of APOE, α2M, and LRP to late-onset AD are particularly intriguing in light of the fact that both apoE and α2M are two key ligands for LRP (5, 6, 8). Both apoE and α2M avidly bind Aβ in vitro and in vivo (5, 35). These observations, together with the finding that LRP and all of its ligands are present in senile plaques (7), strongly implicate the pathogenic importance of the LRP pathway in AD. We found that LRP levels are significantly reduced in AD, compared with healthy controls. Linear-regression analysis revealed that LRP levels progressively decline with the increasing age of control subjects (an inverse correlation) and are further reduced in AD subjects. Among AD patients, however, increased LRP levels were correlated with later age of disease onset, indicating that higher LRP levels might be protective against AD. This apparent protective effect was accentuated among noncarriers of the APOE ε4 allele. As increasing age is the primary risk factor for AD, these data indicate that reduced LRP expression may be one factor contributing to AD susceptibility. This notion is highly consistent with the negative association of the LRP T allele to AD (8, 9, 11, 12) and increased LRP levels among T-allele carriers demonstrated in this study. Although the biological mode of the LRP T allele requires further characterization, we hypothesize that the C766T polymorphism is in linkage disequilibrium with a causative mutation or polymorphism that regulates LRP expression (for example, promoter/enhancer) in the aging brain. ‘’
“In the current study, we provide compelling evidence that Aβ uptake via the α2M-LRP pathway is through a bona fide receptor-mediated mechanism and not through nonspecific Aβ degradation or fluid-phase pinocytosis. –“
“The results of the current study provided the first in vivo evidence of the LRP-clearance pathway in AD pathogenesis. Our observations lead us to postulate that reduced LRP expression is a contributing risk factor for AD, possibly by impeding clearance of soluble Aβ complexes. Functional characterization of α2M polymorphisms associated with AD (6) and future transgenic animal models of LRP and/or α2M expression should further elucidate the mechanism of Aβ clearance and AD pathogenesis. The observation that AD risk or protection associated with LRP levels is strongest among noncarriers of the APOE ε4 allele is particularly interesting in light of the ligand/receptor relationship between apoE and LRP. Because the receptor function of LRP obviously depends on intact activity of its ligands, we hypothesize that high levels of LRP cannot effectively rescue the pathogenic effects of apoE4, the latter operating at a step that negates the clearance mechanism. However, in the presence of apoE2 or apoE3, where the ligand complexes are not perturbed, alterations in LRP level and, in turn, clearance activity become highly consequential for AD pathogenesis. Since increased LRP expression may promote both neuronal survival mediated by apoE2 and apoE3 isoforms and also enhance the clearance of soluble Aβ complexes, the current data provide an alternative direction for AD therapeutic intervention by targeting the Aβ/LRP clearance pathway in non-APOE ε4 carriers.”
3. Haircuts, Tails, and Barbers
APP Image source Amloid beta 40 Image source
Amyloid beta is made up of the cleaved “tail” of the Amyloid precursor protein (APP). APP is a plasma membrane protein that Requires a “haircut”, once APP Trans locates from the ER (protein factory) to the plasma membrane.
The “barbers” that give APP a “haircut” are called “Secretases”. Three “barbers” exist that cut hair at different lengths. The pieces of “hair” cut off APP are the “tails” of the APP molecule and are what Amyloid beta is made up of.
3. It takes two Barbers to cut hair In the brain – each hair must be cut twice!
There are 4 barbers in the brain:
- Alpha-secretase
- Beta-secretase 1 (BACE1)
- Beta-secretase 2 (BACE2) and
- Gamma-secretase
It takes two barbers to cut the hair on the APP protein. If alpha secretase “cuts first” and gamma secretase cuts APP second, no amyloid accumulates in the brain. If BACE “cuts first” and gamma secretase cuts second, then the piece that is produced forms Amyloid aggregates with lengths of 17 to 42 amino acids. The longer pieces of “cut hair” left behind outside of the cell are called Amyloid beta 40 (AB-40) and amyloid beta 42 (Ab-42).
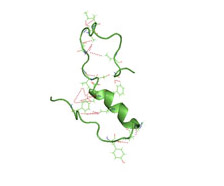 A partially folded structure of amyloid-beta(1 40) in an aqueous environment (pdb 2lfm)[1] Image source
A partially folded structure of amyloid-beta(1 40) in an aqueous environment (pdb 2lfm)[1] Image source
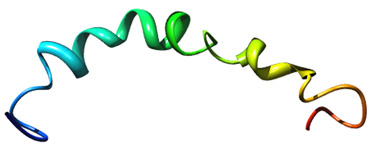 Amloid beta 42 Image source
Amloid beta 42 Image source
AlthoughAB-42 is found in much smaller amounts, compared to the more common AB-40, the AB-42 piece of “cut hair” is much more toxic. The next piece of the puzzle explains why.
4. Why Amyloid beta aggregates: “The salt bridge between Asp23 and Lys28”
Like all polypeptides, Amyloid beta has amino acids with positive and negatively charged side chains. One negatively charged side chain (on aspartate, #23) and one positively charged side chain (on lysine, #28) are attracted to each other and form a “salt bridge”. This salt bridge between Asp23 and Lys28 forces the chain to form a “Hairpin turn,” looping back on itself. (A salt bridge is a bond between a positively charged amino acid side chain and a negatively charged amino acid side chain). The hairpin shape of Abeta makes multiple copies of the polypeptide stick together in parallel, forming a 3D structure called a “beta Pleated sheet”. See the following diagram. This beta-pleated sheet structure is what is referred to as a “Amyloid fibril” and is about 8 nm in size. It is not soluble and starts aggregating outside of the cell.
Ref: Structural conversion of neurotoxic amyloid-β1–42 oligomers to fibrils
5. The “tail of 2 toxicities” – AB42 >> AB-40
Amyloid beta 40 is the common, more soluble form of A-beta. A-beta 42 has two more amino acids on the tail end of the peptide. One of these, amino acid #42, is an alanine, which can loop back to form a salt bridge with amino acid #35, methionine (Met35). This extra hairpin turn of AB-42 makes it less soluble and more toxic. The toxicity of AB-42 is much greater than AB-40. One possible reason for this is described below.
Ref:Figure 1: Sequence and structure of the monomer unit in Aβ40 and Aβ42 fibrils (the following diagram)

“(a) Sequence of Aβ42 that is derived from human APP. (b) Structural constraints in Aβ40 and Aβ42 fibrils. NMR measurements of Aβ40 fibrils have shown that residues 1–10 are unstructured and residues 11–40 adopt a β-turn-β fold19, 20. Side chain packing is observed between Phe19 and Ile32, Leu34 andVal36 and between Gln15 and Val36 as well as between His13 and Val40 (blue dashed lines). In Aβ42 fibrils, residues 1–17 may be unstructured (in gray), with residues 18–42 forming a β-turn-β fold14. Molecular contacts have been reported within the monomer unit of Aβ42 fibrils between Phe19 and Gly38 (red dashed line)14 and between Met35 and Ala42 (orange dashed line)18. In both Aβ40 and Aβ42, the turn conformation is stabilized by hydrophobic interactions (green residues) and by a salt bridge between Asp23 and Lys28 (black dashed line).”
6. Direct neurotoxicity of Abeta
“The mechanism by which amyloid beta may damage and kill neurons is by generating reactive oxygen species during the process of its self-aggregation. When this occurs on the membrane of neurons it causes lipid peroxidation and the generation of a toxic aldehyde called 4-hydroxynonenal which, in turn, impairs the function of ion-motive ATPases, glucose transporters and glutamate transporters. As a result, amyloid beta promotes depolarization of the synaptic membrane, excessive calcium influx and mitochondrial impairment.[51](ref Wikipedia).
“Amyloid-beta(Aβ or Abeta) denotes peptidesof 36–43 amino acidsthat are crucially involved in Alzheimer’s diseaseas the main component of the amyloid plaquesfound in the brains of Alzheimer patients. The peptides result from the amyloid precursor protein(APP), which is being cut by certain enzymes to yield Aβ. Aβ molecules can aggregate to form flexible soluble oligomerswhich may exist in several forms. It is now believed that certain misfolded oligomers (known as “seeds”) can induce other Aβ molecules to also take the misfolded oligomeric form, leading to a chain reaction akin to a prioninfection. The seeds or the resulting amyloid plaques are toxic to nerve cells. The other protein implicated in Alzheimer’s disease, tau protein, also forms such prion-like misfolded oligomers, and there is some evidence that misfolded Aβ can induce tau to misfold.[2] — A recent study suggested that APP and its amyloid potential is of ancient origins, dating as far back as early deuterostomes.[3] (ref Wikipedia).”
7. Abeta and RAGE receptor mediated neurotoxicity: The story of NF-kB, MAPK, and “Inflammaging”
There is a pro-inflammatory membrane receptor found on all cells that binds to “Advanced Glycation End products”, aka AGEs. Hemoglobin A1c is an example of an AGE. The Membrane receptor for AGEs is called the “Receptor for Advanced Glycation Endproducts” or RAGE. Amyloid beta binds to the RAGE receptor and triggers sustained activation if pro-inflammatory pathways including the NF-kB and MAPK pathways. This is especially bad because it induces a prolonged activation of these pathways, whereas most stimuli induce a temporary, short period of pro-inflammatory gene expression.
Although other pathways account for the destruction of Abeta (such as the LDL receptor related protein, or LRP), the RAGE-mediated activation of inflammation in neurons, astrocytes, and glial cells is thought to induce cellular senescence, “inflammaging”, and neuronal cell death by multiple gene products induced by NF- kB and MAPK, such as IL-1b, TNF-a, IL-6, IL-8, ROS, RNS, and other Molecules secreted as part of the SASP. In all of this “inflammaging,” autophagy is also up regulated. Eventually the neurons die by apoptosis or autophagy-induced Cell death (AICD). .
Ref: RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. “Amyloid-beta peptide is central to the pathology of Alzheimer’s disease, because it is neurotoxic–directly by inducing oxidant stress, and indirectly by activating microglia. A specific cell-surface acceptor site that could focus its effects on target cells has been postulated but not identified. Here we present evidence that the ‘receptor for advanced glycation end products’ (RAGE) is such a receptor, and that it mediates effects of the peptide on neurons and microglia. Increased expressing of RAGE in Alzheimer’s disease brain indicates that it is relevant to the pathogenesis of neuronal dysfunction and death). This topic is discussed in the earlier-mentioned blog entries on microglial activation and neurological diseases.
Site-Specific Blockade of RAGE-Vd Prevents Amyloid-β Oligomer Neurotoxicity. “In the genesis of Alzheimer’s disease (AD), converging lines of evidence suggest that amyloid-β peptide (Aβ) triggers a pathogenic cascade leading to neuronal loss. It was long assumed that Aβ had to be assembled into extracellular amyloid fibrils or aggregates to exert its cytotoxic effects. Over the past decade, characterization of soluble oligomeric Aβ species in the brains of AD patients and in transgenic models has raised the possibility that different conformations of Aβ may contribute to AD pathology via different mechanisms. The receptor for advanced glycation end products (RAGE), a member of the Ig superfamily, is a cellular binding site for Aβ. Here, we investigate the role of RAGE in apoptosis induced by distinct well characterized Aβ conformations: Aβ oligomers (AβOs), Aβ fibrils (AβFs), and Aβ aggregates (AβAs). In our in vitrosystem, treatment with polyclonal anti-RAGE antibodies significantly improves SHSY-5Y cell and neuronal survival exposed to either AβOs or AβAs but does not affect AβF toxicity. Interestingly, using site-specific antibodies, we demonstrate that targeting of the Vd domain of RAGE attenuates AβO-induced toxicity in both SHSY-5Y cells and rat cortical neurons, whereas inhibition of AβA-induced apoptosis requires the neutralization of the C1d domain of the receptor. Thus, our data indicate that distinct regions of RAGE are involved in Aβ-induced cellular and neuronal toxicity with respect to the Aβ aggregation state, and they suggest the blockage of particular sites of the receptor as a potential therapeutic strategy to attenuate neuronal death.”)
Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer’s disease: identification of a cellular activation mechanism. “Receptor-mediated interactions with amyloid beta-peptide (Abeta) could be important in the evolution of the inflammatory processes and cellular dysfunction that are prominent in Alzheimer’s disease (AD) pathology. One candidate receptor is the receptor for advanced glycation endproducts (RAGE), which can bind Abeta and transduce signals leading to cellular activation. Data are presented showing a potential mechanism for Abeta activation of microglia that could be mediated by RAGE and macrophage colony-stimulating factor (M-CSF). Using brain tissue from AD and nondemented (ND) individuals, RAGE expression was shown to be present on microglia and neurons of the hippocampus, entorhinal cortex, and superior frontal gyrus. The presence of increased numbers of RAGE-immunoreactive microglia in AD led us to further analyze RAGE-related properties of these cells cultured from AD and ND brains. Direct addition of Abeta(1-42) to the microglia increased their expression of M-CSF. This effect was significantly greater in microglia derived from AD brains compared to those from ND brains. Increased M-CSF secretion was also demonstrated using a cell culture model of plaques whereby microglia were cultured in wells containing focal deposits of immobilized Abeta(1-42). In each case, the Abeta stimulation of M-CSF secretion was significantly blocked by treatment of cultures with anti-RAGE F(ab’)2. Treatment of microglia with anti-RAGE F(ab’)2 also inhibited the chemotactic response of microglia toward Abeta(1-42). Finally, incubation of microglia with M-CSF and Abeta increased expression of RAGE mRNA. These microglia also expressed M-CSF receptor mRNA. These data suggest a positive feedback loop in which Abeta-RAGE-mediated microglial activation enhances expression of M-CSF and RAGE, possibly initiating an ascending spiral of cellular activation.”)
Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease “In this review, we revisit the hypothesis that advanced glycation endproducts (AGEs) and their receptor RAGE may play an important role in disease pathogenesis. Accumulation of AGEs in cells and tissues is a normal feature of aging, but is accelerated in AD. In AD, AGEs can be detected in pathological deposits such as amyloid plaques and neurofibrillary tangles. AGEs explain many of the neuropathological and biochemical features of AD such as extensive protein crosslinking, glial induction of oxidative stress and neuronal cell death. Oxidative stress and AGEs initiate a positive feedback loop, where normal age-related changes develop into a pathophysiological cascade. RAGE and its decoy receptor soluble RAGE, may contribute to or protect against AD pathogenesis by influencing transport of β-amyloid into the brain or by manipulating inflammatory mechanisms. Targeted pharmacological interventions using AGE-inhibitors, RAGE-antagonists, RAGE-antibodies, soluble RAGE or RAGE signalling inhibitors such as membrane-permeable antioxidants may be promising therapeutic strategies to slow down the progression of AD.”
In his treatise ANTI-AGING FIREWALLS – THE SCIENCE AND TECHNOLOGY OF LONGEVITY originally drafted in 2008, Vince discussed the roles of AGE’s as one of the key sources of aging.
- Amyloid beta clearance – the role of ApoE as the “1st clean up protein”
ApoE is a “transport protein” whose primary function in the brain is to help transport Cholesterol and phospholipids in the brain to the sites of new neuron spine formation (called neurites), new neuron cell growth, and myelin formation. The role of ApoE in this transport system is that of a “docking protein”, much like ApoA and ApoB are docking proteins for the HDL and LDL transport systems in the blood that transport cholesterol to the liver (HDL) and away from the liver (LDL).
ApoE has an additional “2nd job” in the brain, however. It is also part if the “clean up crew” for clearing soluble Amyloid beta out if the brain. ApoE first sticks to the hydrophobic portions of the Amyloid beta and then clears the Abeta out of the extra cellular space by binding to the LDL receptor related protein (LRP) on the surface of the brain microglial cells. The reason ApoE4 is associated with an increased risk of AD is explained below in section 10.
9. Abeta clearance by alpha2 macro-globin (a2M) – the 2nd “clean up Protein
There is a second clean-up system for disposing of extracellular junk proteins. This system utilizes a protein called “alpha 2 macro-globin, or a2M. a2M binds to soluble amyloid beta and then binds to LRP, just like ApoE.
10. ApoE4 vs ApoE3 vs ApoE2
Three variants of the ApoE protein are found in the population at large. They vary in sequence at two locations on the ApoE amino acid chain. The 3 forms of the protein are called ApoE2, ApoE3, and ApoE4. ApoE4 has a different binding Affinity for LRP than ApoE3 or ApoE2. This is why ApoE4 does not clear the Amyloid beta as well.
The 2000 publication Modulation of amyloid β-protein clearance and Alzheimer’s disease susceptibility by the LDL receptor–related protein pathway reports: “Susceptibility to Alzheimer’s disease (AD) is governed by multiple genetic factors. Remarkably, the LDL receptor–related protein (LRP) and its ligands, apoE and α2M, are all genetically associated with AD. In this study, we provide evidence for the involvement of the LRP pathway in amyloid deposition through sequestration and removal of soluble amyloid β-protein (Aβ). We demonstrate in vitro that LRP mediates the clearance of both Aβ40 and Aβ42 through a bona fide receptor-mediated uptake mechanism. In vivo, reduced LRP expression is associated with LRP genotypes and is correlated with enhanced soluble Aβ levels and amyloid deposition. Although LRP has been proposed to be a clearance pathway for Aβ, this work provides the first in vivo evidence that the LRP pathway may modulate Aβ deposition and AD susceptibility by regulating the removal of soluble Aβ.”
— “The LDL receptor–related protein (LRP) is a multifunctional receptor that mediates the internalization and degradation of ligands involved in metabolic pathways of lipoproteins and protease/protease-inhibitor complexes (1), including α2-macroglobulin (α2M) (2), apoE (3), and Kunitz protease inhibitor (KPI) containing forms of amyloid precursor protein (APP) (4). Remarkably, the aforementioned ligands are all genetically associated with Alzheimer’s disease (AD) (5, 6) and are found in senile plaques in brains of AD patients (7). To date, the strongest evidence directly implicating a role of LRP in AD is from genetic studies first reported by us (8) and subsequently confirmed in four independent case-control cohorts (9–12). In addition, another genetic polymorphism in LRP was found to be associated with AD (13), further evidence corroborating the LRP gene as an important AD-susceptibility locus. Our study reported a genetic polymorphism (C766T) in exon 3 of LRP that is under-represented in AD and associated with later age of disease onset. However, the underlying biologic relevance of the silent LRP C766T polymorphism is unclear. Moreover, the precise mechanisms by which LRP and its ligands may contribute to AD pathogenesis are unknown.”
— “In the current study, we provide compelling evidence that Aβ uptake via the α2M-LRP pathway is through a bona fide receptor-mediated mechanism and not through nonspecific Aβ degradation or fluid-phase pinocytosis. This was shown by the competition of 125I-Aβ uptake with excess unlabeled Aβ complexes and the complete saturation of 125I-Aβ uptake at physiological concentrations. So far, no other Aβ uptake pathway meets the criteria for a bona fide receptor-mediated mechanism. Although the scavenger receptor has been postulated to mediate the uptake of amyloid fibrils, such process is not subject to competition and saturation of the receptor (30, 31). At another level of Aβ catabolism, recent observations have indicated that neutral endopeptidase and insulin-degrading enzyme are both capable of degrading extracellular Aβ in a cell-autonomous manner (36,37). Thus, it is likely that there are multiple ways of mediating Aβ degradation in vivo. Our study demonstrated that LRP does not alter the secretion of Aβ from APP695-expressing cells but is required for α2M-mediated clearance of soluble Aβ. Because both LRP and APP695 are predominantly expressed in neurons, reduced LRP levels in the AD brain is predicted to negatively impact the clearance of soluble Aβ but not its production in neurons. However, it is important to note that LRP overexpression in LRP-deficient CHO cells results in altered trafficking of KPI containing APP751 (28), possibly through the APP-LRP physical interaction (4). Thus, it is possible that LRP also alters APP trafficking (i.e., internalization/recycling) and Aβ generation through other mechanisms. As APP isoforms are differentially expressed in neurons and glia, LRP-clearance activity versus altered APP trafficking might be differentially modulated across cell types.”
— “The genetic association of LRP T polymorphism with both increased LRP expression and reduced amyloid deposition is intriguing in light of the in vitro evidence of Aβ clearance through the LRP pathway (16, 17). These observations are now further strengthened by genetic association of LRP with soluble Aβ levels in plasma. It is noteworthy that the pathogenic Aβ42 species is as effectively cleared through the LRP pathway as Aβ40 (Figure 1a), an activity that may dramatically impact amyloid deposition in vivo. Accordingly, we interpret these data to indicate that reduced LRP expression, at least in part, contributes to increased Aβ levels and amyloid deposition by negatively impacting Aβ clearance. This interpretation is consistent with our observation that reduced LRP expression is also correlated with increased AD susceptibility and earlier age of disease onset. In our cell-culture system, we demonstrated the requirement of LRP in the α2M-mediated clearance of Aβ. However, it has been reported that apoE and lactoferrin, two other LRP ligands, also sequester Aβ and mediate its clearance (16, 38). Thus, reduced LRP levels may impede the clearance of various Aβ complexes. Interestingly, many LRP ligands, including apoE, α2M, and lactoferrin, are produced from astrocytes, whereas LRP is largely expressed in neurons. Thus, it is likely that receptor-mediated uptake and clearance of soluble Aβ complexes occur in neurons, whereas uptake of fibrillar amyloid is mediated by microglia (30, 31). In this regard, downregulation of LRP expression has been linked to proinflammatory stimuli such as LPS and IFN-γ in cultured cells (39, 40). We speculate that proinflammatory processes present in the AD brain may induce downregulation of LRP expression, further reducing Aβ clearance and enhancing amyloid deposition. Since LRP mediates the normal function of neuronal remodeling through internalization of apoE (19), reduced LRP expression in aging and disease may also compromise neuronal viability independent of the effects on Aβ clearance.”
— “The results of the current study provided the first in vivo evidence of the LRP-clearance pathway in AD pathogenesis. Our observations lead us to postulate that reduced LRP expression is a contributing risk factor for AD, possibly by impeding clearance of soluble Aβ complexes.”
New predictive tests for Alzheimer’s Disease
Over the years a number of tests have been proposed or developed for confirming the existence of AD or predicting the onset of AD in presymptomatic individuals, These tests have ended to be expensive imaging procedures (e.g. PET scan, 3D volumetric MRI, etc), or require invasive procedures (e.g. a CSF test which requires a lumbar puncture) or have relatively low accuracy (e.g. like some of the individual tests for proteins developed in the 1990s and early 2000s mentioned below).
Lipid biomarker test
A lipid biomarker test for AD has been under development at Georgetown and Rochester Universities. As reported in the 2014 story in the ALZFORUM Do Lipids Hold the Key to Blood-Based Alzheimer’s Test? “Researchers have long been searching for a blood-based test that could predict Alzheimer’s disease. In the March 9 Nature Medicine, researchers led by Howard Federoff at Georgetown University Medical Center, Washington, D.C., and Mark Mapstone at the University of Rochester School of Medicine, New York, report that a panel of 10 lipids could one day fit the bill. In a small study, plasma levels of these lipids distinguished, with 90 percent accuracy, who would develop cognitive impairment over the next two to three years. If the results are confirmed in larger samples, it would encourage hopes for a blood test for Alzheimer’s. However, the authors stress that is still years away. Numerous questions remain, including whether the observed lipid changes are specific to AD, or represent a more general marker for neurodegeneration.”
“The ability to identify individuals who are at risk of developing Alzheimer’s before the clinical manifestation of cognitive impairment has long been a Holy Grail of the neuromedicine community,” said Mark Mapstone, Ph.D., a neuropsychologist with the University of Rochester School of Medicine and Dentistry and lead author of the study.“Current efforts to develop a treatment for this disease are coming up short because they are probably being used too late. Biomarkers that can allow us to intervene early in the course of the disease could be a game-changer(ref).” Other criteriia for an effective predictive test for AD is that is should be relatively cheap and non-invasive It is expected that this “lipid bio marker test” will cost around $200.
Combination protein test
Lipids are tricky to measure in blood, however. Proteins, on the other hand, are much easier to measure. A new “protein test” from the European Consortium group looks at 10 plasma proteins, rather than 10 lipids. It has an accuracy of 87% and a specificity of 88%. (Not quite as good, but the test may be easier to do in the lab. The test is described in the July 2014 publication Plasma proteins predict conversion to dementia from prodromal disease. “Background:The study aimed to validate previously discovered plasma biomarkers associated with AD, using a design based on imaging measures as surrogate for disease severity and assess their prognostic value in predicting conversion to dementia. – Methods: Three multicenter cohorts of cognitively healthy elderly, mild cognitive impairment (MCI), and AD participants with standardized clinical assessments and structural neuroimaging measures were used. Twenty-six candidate proteins were quantified in 1148 subjects using multiplex (xMAP) assays. Results: Sixteen proteins correlated with disease severity and cognitive decline. Strongest associations were in the MCI group with a panel of 10 proteins predicting progression to AD (accuracy 87%, sensitivity 85%, and specificity 88%). Conclusions: We have identified 10 plasma proteins strongly associated with disease severity and disease progression. Such markers may be useful for patient selection for clinical trials and assessment of patients with predisease subjective memory complaints.”
Here is the list of 10 proteins from this new “protein test” and my comments on each one, along with some references. It is surprising how many are actually “neuroprotective Proteins” (5), rather than “neurotoxic proteins” (3) and that many have been known for some time.
1. TTR – Transthyretin is a protein that binds to amyloid beta and prevents it from aggregating. Thus it is neuroprotective. In AD, there is lower levels of TTR.
See the 2011 publication Human serum transthyretin levels correlate inversely with Alzheimer’s diseaseand the 2012 publication Plasma transthyretin as a candidate marker for Alzheimer’s disease
2. Clusterin (aka ApoJ) – This is another “clean up crew” protein that cleans up bad (Damaged) lipids in the cell membranes. It is neuroprotective in Alzheimer’s disease.
See the 2012 publication Clusterin in Alzheimer’s disease and the 2012 publication The role of clusterin in Alzheimer’s disease: pathways, pathogenesis, and therapy.
3. Cystatin C – This is the most abundant serine protease inhibitor found in the extra-cellular space. It binds to amyloid beta and prevents it from aggregating and forming sheets of amyloid. Thus, it is Neuroprotective.
See the 2012 publication Cystatin C in Alzheimer’s disease.
4. Alpha 1 acid Glycoprotein – This is an acute phase protein, like C-reactive protein. It is produced in response to inflammation. In sick people in the hospital, it predicts death.
See the 2003 publication Alpha 1-acid glycoprotein is an independent predictor of in-hospital death in the elderly.
5. ICAM1 – This is an intercellular adhesion molecule. It attracts inflammatory cells such as macrophages to the site and holds the cells there (I.e. Makes the site “sticky”). Levels in the brain are increased with AD and it appears to be coming from astrocytes,
See the 1993 publication Expression of intercellular adhesion molecule (ICAM)-1 by a subset of astrocytes in Alzheimer disease and some other degenerative neurological disorders and the 1991 publication Expression of intercellular adhesion molecule 1 (ICAM-1) in Alzheimer’s disease.
6. CC4 – complement C4 – This molecule is part of the classic pathway of the complement system. Levels of this are elevated in the brain with AD.
See the 2012 publication Cerebrospinal fluid levels of complement proteins C3, C4 and CR1 in Alzheimer’s disease.
7. PDEF – Pigment epithelium derived factor. This also protects nerve cells from death.
See the 2002 publication Pigment Epithelium-Derived Factor Protects Cultured Cerebellar Granule Cells Against Glutamate-Induced Neurotoxicity
8. A1AT – alpha 1 antitrypsin – This is a protease inhibitor made by astrocytes. It is found in the Abeta plaques.
See the 1994 publication Serum alpha 1-antitrypsin and alpha 2-macroglobulin in Alzheimer’s and Binswanger’s disease. And the 1992 publication [alpha]1-Antitrypsin and [alpha]1-antichymotrypsin are in the lesions of Alzheimer’s disease.
9. RANTES (ApoC3) – This appears to be a “cellular stress resistance protein”. When it is secreted in cases of AD, it protects the nerve cells from dying
See the 2010 publication RANTES upregulation in the Alzheimer’s disease brain: a possible neuroprotective role.
10. ApoE genotype – This is obviously a good predictor of earlier onset of LOAD.
More on Alzheimer’s disease
There is much more to the AD story besides the beta-amloid aspects and others discussed here. Jim has more to say. previous blog entries are mentioned above, and Vince has been working for some time on a blog on the tau tangles aspects of AD and other neurological diseases. So, stand by for more!

{kind=link}
{kind=link}
{kind=link}
Pingback: A simple, comprehensive plan to prevent or reverse Alzheimer’s Disease and other neurodegenerative diseases – Part 1: The Plan | AGING SCIENCES – Anti-Aging Firewalls