
By James P Watson, with editorial assist, a little rounding out and a few comments by Vince Giuliano

I am convinced that actions in the Renin-Angiotensin cascade can be a major cause of inflammation, aging, and cancer. Exogenous antioxidants, Bucky balls, and polyphenols can do little if anything to stop actions in this pathway which can be major drivers of hypertension, heart disease, stroke, cancer, and aging. Actions in this pathway may explain why all of the clinical trials to date of polyphenols such as curcumin, ECGC, resveratrol, isothiocyanates, Vitamin E, beta-carotene, etc. have failed or display mixed results. We must learn to understand this system better.
 Image source Human angiotensin
Image source Human angiotensin

Image source Angiotensin converting enzyme (ACE)
Introduction
The molecular story of life has over 300 major “actors” that talk the language of intercellular communication. Some of these “actors” only speak one language. Most play the “hero” or the “villain” but not both. No single “actor” can fit into all of the acting roles required for intercellular communication, but if there were some “actors who could play multiple roles”, the best two would be Angiotensin Converting Enzyme (ACE) and Angiotensin II (ANG). These two molecules can play 10 roles. They also are “double agents” who can work for the enemy and the “good guys” at the same time. The roles appear to be very important and quite diverse, for example, assisting a sperm to find an egg during conception.
Specifically, Angiotensin II can play the role of a hormone, a neurotransmitter, a cytokine, and a growth factor. ANG (Angiotensin II) can make blood clot or prevent blood from clotting. ACE can cause systemic hypertension or can cause a local plaque to form in a specific blood vessel location. Working together, ACE and ANG can cause vasodilation or vasoconstriction, depending on which Angiotensin receptors are present on the target cell surface. Likewise, ACE and ANG can exert a pro-inflammatory or anti-inflammatory effect in the immune system, depending on which Angiotensin recptors are present in the target immune cells. The double roles are reminiscent of the double roles of oxidative stress and free radicals – they can do good or harm. ACE and ANG increases oxidative stress in the vascular system and can therefore accelerate aging and hardening of the arteries via one receptor type on the target cell, but can do the opposite with another receptor type. ACE and ANG can increase oxidative stress in other tissues and drive cancer formation via one receptor type, but in other cells, the pair can trigger an anti-oxidant response. As the cartoon dog Peanuts says, ARGH!!!
As a result, these two “actors” appear to be “Double Agents” in the Molecular Story of Life. Here is a list of all of the ways that this pair of molecules can signal:
1. Hormone – ACE and ANG circulate in the plasma and exert effects at distant sites via 7 different receptors (AT1, 2, 3, etc.).
2. Neurotransmitter – ANG is a neurotransmitter in the paraventricular nucleus of the hypothalamus, activating AT1 receptors.
3. A growth factor – ANG is a key growth factor in skin wound healing via AT1R (controls keratinocyte & fibroblast migration).
4. An angiogenesis factor – ANG induces expression of VEGF and induces angiogenesis via AT2R, but requires hypoxia, bradykinin, the B2 bradykinin receptor, and nitric oxide .
5. A vascoconstrictor and a vasodilator– ACE and ANG exert a potent vasoconstrictive effect via ATR1 receptors, but have a vasodilator effect via the ATR2-7 receptors. Thus their effects are “cell-specific” and “tissue-specific.”
6. A pro-thrombotic agent – ANG stimulates thrombosis via inducing expression of the thrombin receptor, release of PAF (platelet activating factor), or by stimulating platelet aggregation and adhesion.
7. A cytokine – ANG stimulates cell proliferation via Akt and MAPK pathways (ERK1/2, p38, and JNK).
8. A driver of aging and inflammation – ANG can chronically up regulate reactive oxygen species via AP-1 and NF-kB.
9. An anti-inflammatory pathway – ACE and ANG can have an anti-inflammation, and anti-oxidative effects via the AT2 receptor.
Molecular Biology of Angiotensin II (ANG)
ANG is made by angiotensin converting enzyme (ACE) which comes in two forms: testicular ACE (tACE) and somatic ACE (sACE). Angiotensin II works primarily via the angiotensin 1 receptor (ATR1) and the angiotensin 2 receptor (ATR2), but there are actually 7 AT receptors in humans. ATR1 and ATR2 have almost diametrically opposed effects. For instance, ATR1 induces vasoconstriction, whereas ATR2 induces vasodilation. ANG induces ROS via the AT1 receptor, but induces anti-oxidant effects via AT2 receptor. ANG increases growth via the ATR1, but decreases growth via ATR5-7. Thus, Angiotensin II is a pleiotrophic factor for many reasons. Here are a few diagrams showing of how ANG and ACE work.

Image source: “Overview of the renin-angiotensin system (RAS). AGT indicates angiotensinogen; Ang, angiotensin; ACE, angiotensin-converting enzyme; ACE2, angiotensin-converting enzyme 2; AT1R, angiotensin type 1 receptor; AT2R, angiotensin type 2 receptor; MasR, Mas receptor.“
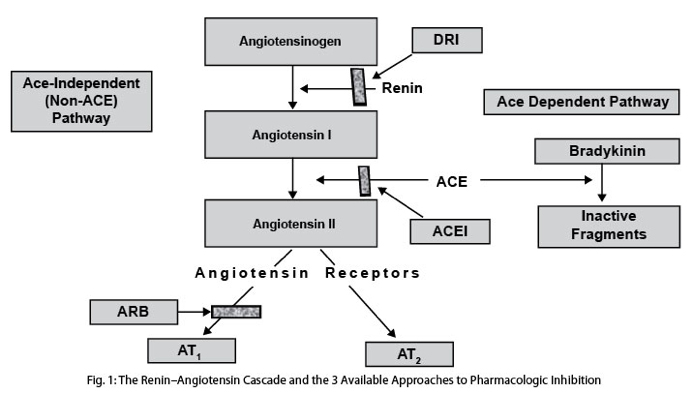
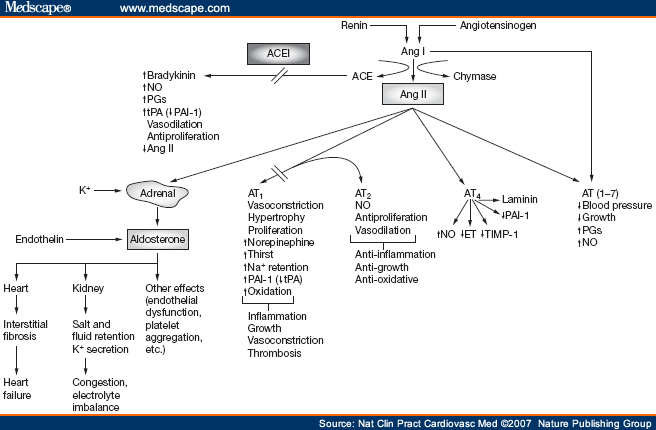
Picture source Some receptor-mediated effects of angiotensins 1 and 2
1. CO, NO, H2S, Prostacyclin (PGI2), and Endocanabanoids are biological antagonists of Angiotensin II
Whereas angiotensin II promotes vasoconstriction and thrombosis, there are several antagonistic pathways that counteract this. In the past, prostacyclin (PGI2) produced by cyclooxygenase-1 (COX-1) and nitric oxide (NO) produced by endothelial nitric oxide synthetase (eNOS) were the only substances known to counteract Angiotensin II.
Now we know of at least 5 major substances that counteract (antagonize) the effects of Angiotensin II as follows:
1. PGI2 => PGI2 works via a smooth muscle cell 2nd messenger, cAMP
2. NO – NO works via a smooth muscle cell 2nd messenger, cGMP.
3. H2S – H2S works via a smooth muscle potasium ATP channel, Katp
4. CO – CO works via a smooth muscle cell 2nd messenger, cGMP
5. Endocanabanoids – Anandamide works via a smooth muscle cell receptor, CB1R
Now we know that both carbon monoxide (CO and hydrogen sulfide (H2S) are “endothelial-derived relaxing factors” (EDRF). H2S is made from L-cysteine by the endothelial enzyme cystathionine gamma-lyase (CSE) and works via K+atp channel and endothelial hyper polarizing factor (EDHF). CO is made from heme breakdown via the enzymes heme oxygenase-1 and 2 (HO-1, HO-2) and work via a cGMP 2nd messenger.
The following diagrams are characterizations of factors involved in vasodialation and vasoconstriction including angiotensin:
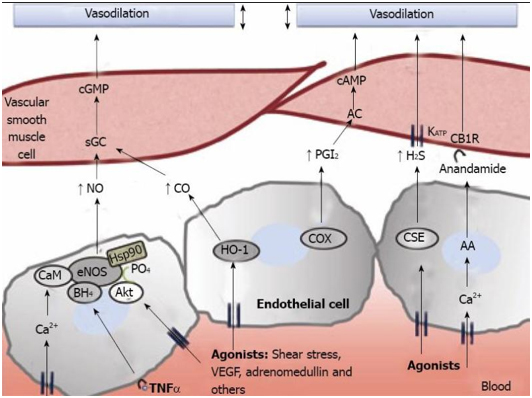
Picture and legend reference: Physiopathology of splanchnic vasodilation in portal hypertension
“Figure 2 Molecular pathways associated to splanchnic vasodilation. Vasoactive molecules involved in the regulation of vascular tone in the arteries of the splanchnic circulation. Nitric oxide (NO), carbon monoxide (CO), prostacyclin (PGl2) or hydrogen sulfide (H2S), generated through different pathways in endothelial cells, cause vasodilation in vascular smooth muscle cells by either activating soluble guanylate cyclase (sGC) to generate cyclic guanosine monophosphate (cGMP), by stimulating adenylate cyclase (AC) and generation of cyclic adenosine monophosphate (cAMP) or through the opening of KATP channels. Also, anandamide activates endothelial cannabinoid 1 receptors (CB1R) provoking vasodilation. AA: arachidonic acid; AC: adenylyl cyclase; Akt: protein kinase B; BH4: tetrahydrobiopterin; CaM: calmodulin; CSE: cystathionine-γ-lyase; COX: cyclooxygenase; eNOS: endothelial nitric oxide synthase; HSP90: heat shock protein 90; IP3: inositol triphosphate; TNFα: tumor necrosis factor α; VEGF: vascular endothelial growth factor.”

Picture source: Hydrogen sulfide: a new EDRF “Molecular basis for endothelium-dependent vasorelaxation. With different substrates, NO, H2S, and PGI2 are produced from the endothelium by NOS, CSE, and COX-1, respectively. They are released from the endothelium and act on the adjacent smooth muscle cells to induce relaxation. NO and H2S are also produced in smooth muscle cells. For brevity of description, smooth muscle production of these substances is not included in this diagram. NOS, NO synthase; CSE, cystathionine -lyase; COX-1, cyclooxygenase-1; AA, arachidonic acid; EDHF, endothelium-derived hyperpolarizing factor.”

picture reference: http://district.bluegrass.kctcs.edu/ron.wasielewski/110studentAps/regulationOfVascularTone-color.jpg
2. Angiotensin II is a very strong inducer of ROS which explains the role of angiotensin in cancer development. Such ROS can be inhibited by the polyphenol caffeoylquinic acid (CQC)
Angiotensin II may be the greatest “driver” of systemic ROS as seen in aging, cancer, and disease. Angiotensin II induces the transcription of several protein components of NADH oxidase and NADPH oxidase. NOX produces a lot of superoxide and are bound just below the plasma membrane of the following types of cells:
- endothelial cells,
- vascular smooth muscle cells,
- adventitial fibroblasts.
NOX produce a lot of superoxide in the phagosomes of the following types of cells:
- neutrophils,
- macrophages.
The downstream targets of this ROS include activation of all of the Redox sensitive kinase pathways to induce many genes via NF-kB and AP-1. The targets of ROS activation are:
A. Akt – this is a potent driver of cancer,
B. The 3 MAPKs:
1. JNK,
2. ERK ½,
3. p38.
These pathways can induce cell proliferation, hypertrophy, and cell migration.
About CQC
The polyphenol from the Taiwanese folk medicine plant, Elephantopus mollis, called 3,4-di-O-caffeoylquinic acid (CQC) can do the following:
1. Inhibits the Angiotensin II-induced proliferation of vascular smooth muscle
2. Counteract this ROS-induced endothelial cell stress
3. Minimizes the ROS-induced impairment of EDRF induced blood vessel relaxation
4. Prevents the Cu++-induced oxidation of human LDL
5. Acts as an antioxidant

Picture source: “Figure 6: Proposed molecular target of CQC to inhibit Ang-II-induced proliferation and/or migration of rVSMC. Arrows indicate the main biological end points preceding cell proliferation and migration in response to Ang II. ROS activate hypertrophy and proliferation in VSMC. In response to the growth factors Ang II or PDGF, ROS are endogenously produced by VSMC and stimulate MAPKs and Akt. Our data documented that CQC inhibited Ang-II-induced rVSMC proliferation and migration. The anti-atherosclerotic property of CQC may act by downregulating the Akt, JNK and in part of ERK1/2 pathways by Ang II. AT1: angiotensin type 1 receptor; GFR: growth factor receptor; Gq: Gq protein.”
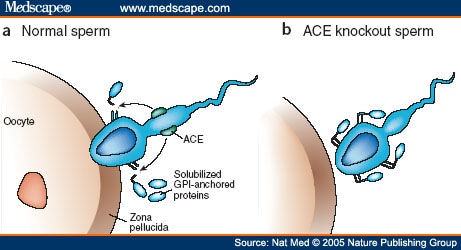
3. During Evolution, the Angiotensin Converting Enzyme (ACE) underwent duplication at the origin of Chordata, just before the “Cambrian Explosion” in a number of species. The ancestral, unduplicated form still exists in the testes, tACE, and is used by sperm for navigation to the egg.
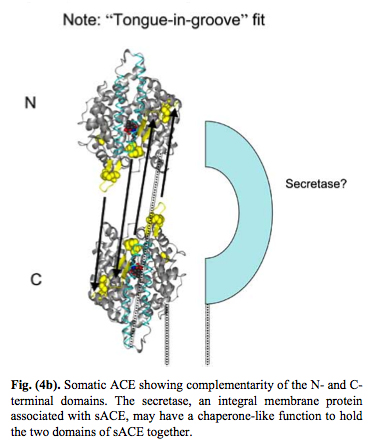
As a result of evolution, the ACE enzyme underwent duplication and now forms a plasma membrane dimer where the two pieces fit together in a “tongue-and-grove” fashion. Thus, the somatic form of the Angiotensin Converting Enzyme (sACE) exists on plasma membranes as a dimer. Here are some illustrations of this:
Reference for next two images: The Central Role of Angiotensin I-Converting Enzyme in Vertebrate Pathophysiology
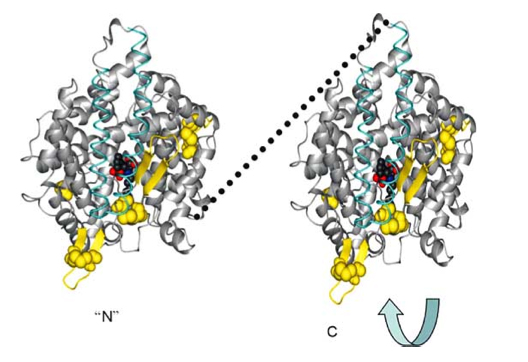
sACE
The unduplicated ancestral version of ACE still exists in the testes. It did not undergo evolution. Here is a picture of the testicular form of ACE:
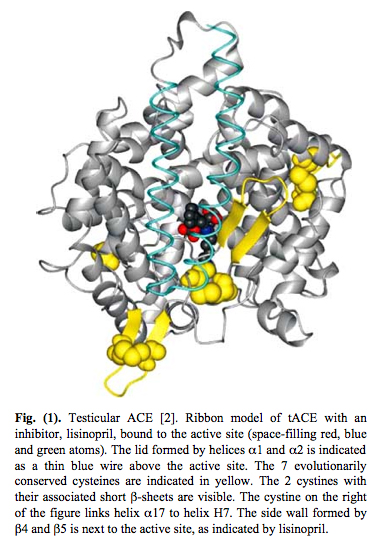
4. Testicular ACE is a Redox Sensor and a Mechanosensor – It is used by sperm as a “GPS” for navigation from the testes to the fallopian tube. Here is an illustration of how important testicular ACE is as a mechanoreceptor on sperm:
The key physical feature that led to figuring out that Testicular ACE is a redox sensor is that it has 7 evolutionarily conserved cysteines. Six of these cysteines form disulfide bonds to nearby intramolecular cysteine residues. These 6 cysteines form a “disulfide bond zipper” for the lid on the active pocket. One cysteine acts as a “latch” for the pocket. These 7 evolutionarily conserved cysteines are depicted in yellow in the diagram on Testicular ACE above. Testicular ACE appears to mainly exist in the oxidized form, which is required for disulfide bond formation. Testicular ACE is very resistant to even exhaustive redox reduction. A 900-fold redox reduction of tACE only decreased the activity of tACE by 78%. Thus it appears that the oxidized form is the active form with the disulfide bonds intact.
Testicular ACE has an active site with a “lid” on it. This allows it to function as a mechanosensor. Here is a picture of this:
5. Somatic ACE (sACE) is a Redox Sensor and exists in a dimeric form in the plasma membrane or as a soluble dimer in the plasma. The plasma form is active in the oxidized state and exists with 90% of plasma sACE oxidized and 10% of plasma sACE reduced.
If the monomeric tACE is a “redox sensor” equivalent to a gun, then the dimeric sACE is a “redox sensor” equivalent to a double-barrel shot gun. With double the number of redox sensitive cysteines, there can be a much greater ability to be oxidized or to absorb reducing agents. Two electrons could start the entire side walls of both active sites “flapping”. Thus, the “disulfide zipper” on sACE is twice as long as the “disulfide zipper” on tACE.
Here is an illustration of this effect:

Illustration reference: The Central Role of Angiotensin I-Converting Enzyme in Vertebrate Pathophysiology
Normally Somatic ACE (sACE) exists as a plasma membrane protein, but it can also be cleaved by a plasma membrane secretase called “ACE secretase” and then it can become a soluble protein found in the plasma. This process is called “ACE shedding” and is comparable to the amyloid-B shedding seen with Alzheimer’s disease. Here is an illustration of this ACE secretase:
![Epitope mapping of human sACE using EM-based model.Model of human sACE was generated using an EM-based model of porcine sACE [49]. Epitopes localization (colored spots) were taken from the following publications [40], [51]–[55].. Relevant amino acid residues in the ectodomain and juxtamembrane region of ACE and a cartoon of the membrane-bound ACE secretase are indicated: Leu1 and Asp616-N terminal residues of the N- and C-domains, respectively; Pro1193 –the last amino acid residues seen in the 3D structure of C domain; Pro1199 – localization of P1199L mutation causing 5-fold increase in blood ACE levels [32]; Arg1203-C-terminal amino acid residue of soluble ACE [23]; Val1228-Ser1248-transmembrane domain [65]; Ser1277-C-terminal amino acid residue of full-length somatic ACE [17].](http://openi.nlm.nih.gov/imgs/512/278/3187827/3187827_pone.0025952.g005.png)
Illustration reference: An Angiotensin I-Converting Enzyme Mutation (Y465D) Causes a Dramatic Increase in Blood ACE via Accelerated ACE Shedding
“Background: Angiotensin I-converting enzyme (ACE) metabolizes a range of peptidic substrates and plays a key role in blood pressure regulation and vascular remodeling. Thus, elevated ACE levels may be associated with an increased risk for different cardiovascular or respiratory diseases. Previously, a striking familial elevation in blood ACE was explained by mutations in the ACE juxtamembrane region that enhanced the cleavage-secretion process. Recently, we found a family whose affected members had a 6-fold increase in blood ACE and a Tyr465Asp (Y465D) substitution, distal to the stalk region, in the N domain of ACE. — Methodology/principal findings: HEK and CHO cells expressing mutant (Tyr465Asp) ACE demonstrate a 3- and 8-fold increase, respectively, in the rate of ACE shedding compared to wild-type ACE. Conformational fingerprinting of mutant ACE demonstrated dramatic changes in ACE conformation in several different epitopes of ACE. Cell ELISA carried out on CHO-ACE cells also demonstrated significant changes in local ACE conformation, particularly proximal to the stalk region. However, the cleavage site of the mutant ACE–between Arg1203 and Ser1204–was the same as that of WT ACE. The Y465D substitution is localized in the interface of the N-domain dimer (from the crystal structure) and abolishes a hydrogen bond between Tyr465 in one monomer and Asp462 in another. — Conclusions/significance: The Y465D substitution results in dramatic increase in the rate of ACE shedding and is associated with significant local conformational changes in ACE. These changes could result in increased ACE dimerization and accessibility of the stalk region or the entire sACE, thus increasing the rate of cleavage by the putative ACE secretase (sheddase).”
As plasma levels of ACE become higher and higher, this is associated with worsening of a person’s hypertension. A family was recently found that had a 6-fold increase in soluble, plasma ACE levels. This was found to be due to a Tyr465Asp (Y465D) substitution, distal to the stalk region in the N-domain.
Below is an illustration of another gene variant that increased shedding of ACE into the plasma. The enzyme that cleaves ACE is called a “sheddase.” Increased levels of ACE in plasma are associated with higher incidences and more severe forms of hypertension.

6. Increased Shedding of ACE is associated with diseases where Angiotensin II is activated. Shedding can be inhibited with Zinc chelation therapy and increased with Antibodies against specific epitopes of sACE.
(“An epitope, also known as antigenic determinant, is the part of an antigen that is recognized by the immune system, specifically by antibodies, B cells, or T cells(ref).” The exact enzyme (sheddase) that cleaves membrane bound sACE has not yet been characterized. It is known that the soluble, circulating ACE found in the plasma comes from endothelial cells It is believed that many different enzymes can do this, including calpains, Furin, and proteasomes. It is known that such increased shedding is key in the development of the diseases associated with angiotensin II.
Reference: Epitope-specific antibody-induced cleavage of angiotensin-converting enzyme from the cell surface The N domain of somatic angiotensin-converting enzyme negatively regulates ectodomain shedding and catalytic activity.
7. sACE can be directly inactivated by nitric oxide (NO) via S-nitrosylation with 4 of the cysteine residues on the “disulfide zipper.”
Essentially this meant that it is possible to “turn off” ACE with nitric oxide “coating” of the cysteine thiols on the ACE molecule. Remember that you must start with the reduced form of sACE to do this and that 90% of the thiols on sACE are normally oxidized. Thus, it is only the 10% of sACE in plasma that could undergo this S-nitrosylation of the cysteine thiols on the disulfide zipper.
Conclusion:
- Angiotensin II and NO are “biological antagonists”. Angiotensin vasoconstricts and NO vasodialates.
- Angiotensin stimulates expression of all 3 isoforms of NOS (eNOS, iNOS, nNOS) to increase NO.
- NO inactivates the reduced form of sACE via S-nitrosylation of the four central residues in the “zipper” in soluble, reduced form of sACE.
- ROS generated by xanthine oxidase, NADPH and NADH in response to angiotensin degrade NO and diminish signaling.
- This ROS-induced degradation of NO in response to angiotensin signaling is an important target for treating hypertension. This appears to be a “critical center” of hypertension.

Picture source” Dr. Caballero: A fine balance between vasoconstrictors, shown on the left, and vasodilators, shown on the right, regulates vascular tone at every moment. Angiotensin II is a very potent vasoconstrictor that also promotes thrombosis and inflammation. Although angiotensin II is not produced in the endothelium, the enzyme that activates it, known as angiotensin-converting enzyme (ACE) is widely found in endothelial cells. The opposing forces, primarily led by nitric oxide, counterbalance the effects of angiotensin II and other vasoconstrictors. Nitric oxide is one of the most important molecules, as it is the prime vasodilator and anti-inflammatory substance released by the endothelium. Prothrombotic and proinflammatory changes occur as a result of increased angiotensin II activity and/or decreased nitric oxide production or activity.”

8. Angiotensin II is the most important “on switch” for the EPO gene and hemoglobin is actually the “oxygen sensor” for induction of the EPO gene via the serial interplay of NO with the Hb molecule and then the AE1 molecule, then albumin, and then finally sACE. Thus, sACE can be a Renal “critimeter”
Human EPO (erythropoietin) is “a glycoprotein hormone that controls erythropoiesis, or red blood cell production. It is a cytokine (protein signaling molecule) for erythrocyte (red blood cell) precursors in the bone marrow(ref).” This is another amazing property of Angiotensin II and of sACE. I knew that hypoxia “turned on” the gene for EPO directly via HIF-1alpha. They have found that Angiotensin II “turns on” HIF-1a several fold stronger than hypoxia does. The way this works is that oxygenation of hemoglobin displaces NO from the heme groups in hemoglobin. This results in these NO molecules forming S-nitrosylation of the hemoglobin cysteine thiol groups. These Hb-bound NO molecules are then gradually transferred to an erythrocyte “anion exchanger” molecule called AE1, via S-nitrosylation, which is found in high concentrations within the cell. The cytoplasmic tail of this membrane-bound molecule has lots of cysteine residues in it. These NO molecules can then be transferred from AE1 in the erythrocyte to albumin in the plasma, which also has cysteine thiols on it. These can also form S-nitrosylation with NO.
Finally, the NO on albumin can be transferred to sACE in the plasma, thereby inactivating it. This is the story of how “hemoglobin is the oxygen sensor” and sACE is the renal “crimiter.” Here is the reaction:
Reference: The Central Role of Angiotensin I-Converting Enzyme in Vertebrate Pathophysiology

9. sACE plays a major role in the pathology of Diabetes, Gout, Sepsis, DIC, Malignant Hypertension, Sickle Cell Disease, and Pre-eclampsia
I find this to be very fascinating, again indicative of the multiple and very important impacts associated with angiotensins and their conversion inhibition. This point is covered in the 2004 review article The Central Role of Angiotensin I-Converting Enzyme in Vertebrate Pathophysiology. “Genomic epidemiologic data, increasingly supported by clinical outcomes results, strongly suggest that overactivity of angiotensin I-converting enzyme (ACE) may underlie most age-related diseases. Angiotensin II, the main product of ACE, is a pleiotropic hormone, capable of serving as a neurotransmitter, growth factor, angiogenesis factor, vasoconstrictor, pro-thrombotic agent, and cytokine. So it is perhaps not surprising that the ACE D/D genotype is associated with several major psychiatric diseases, most cancers except prostate cancer (where the D/D genotype is actually protective), most cardiovascular diseases, most autoimmune diseases, and even infectious diseases like tuberculosis and HIV. In a preliminary study, angiotensin II blockade appeared to hasten recovery from West Nile virus encephalitis; it may be equally useful in SARS. The ACE gene underwent duplication at the origin of Chordata, just before the “Cambrian Explosion” in the number of species. The ancestral, unduplicated form of ACE is still expressed during the terminal differentiation of human spermatocytes, suggesting a critical role in reproduction. The crystal structure of testicular ACE (tACE) was recently published. Computer modeling suggests that tACE may be activated by both mechanical forces and reducing agents. The duplicated form of ACE (somatic ACE, sACE) is expressed in areas of high fluid flow. sACE may auto-dimerize via a novel protein motif, the “disulfide zipper.” The sACE dimer is predicted to have higher catalytic efficiency and redox resistance than tACE.”
Angiotensin II and sACE in disease processes and aging
1. sACE plays a major role in Inflammation and Autoimmunity
Angiotensin II functions as a cytokine in invertebrates. In vertebrates, it is a pyrogen and causes a fever. sACE appears on the plasma membranes of activated macrophages and T lymphocytes when they are “angry” – the sACE is called “CD143.” T-cells can stimulate the expression of sACE on monocytes in a MHC-restricted manner and vice versa. This involves cell-to-cell contact. Here is a depiction of how this works on a molecular basis:

Reference: again The Central Role of Angiotensin I-Converting Enzyme in Vertebrate Pathophysiology
A. Bacterial infections and Angiotensin II
- Bacterial infections also involve ACE.
- Bacterla infections lower tissue oxygen tension due to the bacterial consumption of oxygen.
- Bacteria also produce lactic acid, lowering tissue pH and causing the pain that a patient typically has with an infection of a lactic acid-producing bacteria (Exception: urinary tract infections that produce urea).
- Under the reducing conditions of hypoxia and low pH, the sACE on the surface of macrophages or T cells becomes activated by the mechanism just diagramed above..
- B. Viral infections and Angiotensin II
- Angiotensin II stimulates the production of the antiviral protein interferon gamma from T cells.
- This interferes with viral replication.
- Angiotensin also helps promote apoptosis in the virally infected cells.
C. Autoimmune Diseae
- Angiotensin is a potent cytokine capable of stimulating the synthesis of other cytokines like MIF, TNF-a, MCP-1, and TGF-b.
- Overactivity of ACE has a been associated with autoimmune diseases such as rheumatoid arthritis, SLE, and fibromyalgia/chronic fatigue syndrome.
- ACE inhibitors have shown clinical responses in patients with psoriasis and alopecia areata.
- D. HIV
- HIV infectivity and progression to AIDS are also associate with the ACE D/D genotype.
- E. Tuberculosis and Leprosy
- Angiotensin blockade may be beneficial in TB as well and with Leprosy
Reference: again, The Central Role of Angiotensin I-Converting Enzyme in Vertebrate Pathophysiology
2. sACE plays a major role in Cancer (with one exception)
- Overactivity of sACE is associated with all solid and hematogenous cancers except prostate cancer in white men, in which sACE activity is actually protective.
- Vascular signaling by sACE thus appears to drive most cancers.
Colon cancer
- For example, many solid cancers, including colon, have recently been shown to be initiated by Wnt, which acts upstream of beta-catenin and APC.
- Wnt is activated by PKC, which is activated by Angiotensin II.
- Thus, Angiotensin II production by vascular endothelial cells (sACE) is an “up stream” cause of increased Wnt signaling.
Breast cancer
- Breast cancer has recently been associated with the ACE D/D genotype in Chinese women.
- Since angiotensin II promotes cell proliferation and angiogenesis, ACE inhibitors or ARBs may be useful adjunctive tx for these D/D genotype of breast cancer.
Pancreatic cancer
- The article referenced here provides an example case of how pancreatic cancer growth can be attenuated with an ACE inhibitor.
Prostate Cancer
- Prostate cancer is the “exception to the rule” for ACE, but only for while men with the D/D genotype.
- In black men, ACE overactivity is associated with prostate cancer as with other cancers.
- In while men with the ACE D/D genotype, ACE overactivity is associated with prostatic hyperplasia but negatively associated with prostate cancer. The reason for this is that sACE is highly expressed in the glandular epithelium of BPH.
- Taking an ACE inhibitor will actually make these prostate cancers in white men grow. The diagram below explains how this works.

reference: yet-again, The Central Role of Angiotensin I-Converting Enzyme in Vertebrate Pathophysiology
3. sACE plays a major role in Neurodegenerative Diseases
Angiotensin II induces the “constitutive increase in ROS” via AP-1. ROS is thought to contribute to the pathogenesis of Alzheimer’s disease, Parkinsons’s disease, Huntington’s disease, ALS, etc, even though we do not think that this is the primary cause of the disease. In other words, ACE inhibition could be a “disease management” approach, rather than a “disease cure” approach. Although we do not think that ACE inhibition alone would prevent any of these diseases, there is good evidence that such would reduce or delay the onset of these diseases.
Reference: yet-again The Central Role of Angiotensin I-Converting Enzyme in Vertebrate Pathophysiology
4. Angiotensin II plays a major role in coronary artery disease
According to the publication Coronary artery superoxide production and nox isoform expression in human coronary artery disease, “Oxidative stress plays important role in the pathogenesis of atherosclerosis and coronary artery disease (CAD). We aimed to determine the sources and selected molecular mechanisms of oxidative stress in CAD – Conclusions: Increased expression and activity of NAD(P)H oxidase subunits and xanthine oxidase, in part mediated through angiotensin II and PKC-dependent pathways, are important mechanisms underlying increased oxidative stress in human coronary artery disease.”
5. sACE plays a major role in Aging
ROS and Chronic Angiotensin II overstimulation
- Angiotensin II stimulates mitochondrial electron transport, oxygen consumption, and the production of ROS;
- Chronic angiotensin II signaling leads to mitochondrial hypertrophy and proliferation..
- This is good, but ongoing chronic overstimulation of Antiotensin II leads to mitochondrial dysfunction with increased uncoupling of electron transport form ATP synthesis and increased production of mitochondrial ROS.
- Inhibition of ACE in old animals restore normal mitochondrial function and gets rid of this mitochondrial uncoupling
The following two diagrams and legends are from Aging and the Renin-Angiotensin System.

Image source “Chronic activation of the renin-angiotensin system (RAS) plays a role in promoting end-stage organ damage associated with aging, by virtue of the ability of angiotensin II (Ang II) to increase tissue reactive oxygen species (ROS) production. ROS compromise mitochondrial integrity and function, leading to a decreased mitochondrial ATP generation. Release of ROS by mitochondria initiates a vicious cycle of progressively increasing oxidative stress with major deleterious effects on numerous organs, including kidney, heart, and brain.”

Image source “Disruption of the angiotensin II type 1a (AT1A) receptor upregulates SIRT3 expression, which suppresses mitochondrial reactive oxygen species (ROS) and prevents mitochondrial damage. Sirt3 deacetylates glutamate dehydrogenase (GDH), isocitrate dehydrogenase 2 (IDH2), acetyl CoA synthase 2 (ACS2), ornithine transcarbamoylase (OTC), and long-chain acyl-CoA dehydrogenase (LCAD). Sirt3 protects against ROS by enhancing the activity of manganese superoxide dismutase (MnSOD) in the mitochondrial matrix and exerts antiapoptotic effects by deacetylating Ku70.”
6. Angiotensin II causes Skeletal Muscle Wasting and Cachexia
- An early part of aging is skeletal muscle wasting (sarcopenia) and cachexia
- Skeletal muscle wasting can be due to angiotensin II, which opposes the action of IGF-1
- Cachexia is due to high circulating levels of TNF-a in the blood. the TNF-a is derived from monocyte/macrophages stimulated by Angiotensin II.
- ACE inhibitors can block this.
Reference: The Central Role of Angiotensin I-Converting Enzyme in Vertebrate Pathophysiology
Final comment (Vince)
It would be nice if we could wrap this all up with a few generalizations that go beyond “The angiotensins and ACE appear to be very important for health and exercise multiple functions in biology.” However. it appears that Angiotensin II is “a walking contradiction, partially saint and (possibly) partially partially ficti0n.” It is a sinner for the most part but not always. Similarly ACE inhibition is often a saintly act but not always, and can also be a sin. And these can be both saints and sinners at the same time. They are double agents and the devil seems always to be in the details. Perhaps we can make more systematic sense of them in the future

Vince
I thought his news from Tel Aviv University was of interest. Erc
Turning Off the “Aging Genes”
Thursday, January 2, 2014
Computer algorithm developed by TAU researchers identifies genes that could be transformed to stop the aging process
Restricting calorie consumption is one of the few proven ways to combat aging. Though the underlying mechanism is unknown, calorie restriction has been shown to prolong lifespan in yeast, worms, flies, monkeys, and, in some studies, humans.
Now Keren Yizhak, a doctoral student in Prof. Eytan Ruppin’s laboratory at Tel Aviv University’s Blavatnik School of Computer Science, and her colleagues have developed a computer algorithm that predicts which genes can be “turned off” to create the same anti-aging effect as calorie restriction. The findings, reported in Nature Communications, could lead to the development of new drugs to treat aging. Researchers from Bar-Ilan University collaborated on the research.
“Most algorithms try to find drug targets that kill cells to treat cancer or bacterial infections,” says Yizhak. “Our algorithm is the first in our field to look for drug targets not to kill cells, but to transform them from a diseased state into a healthy one.”
A digital laboratory
Prof. Ruppin’s lab is a leader in the growing field of genome-scale metabolic modeling or GSMMs. Using mathematical equations and computers, GSMMs describe the metabolism, or life-sustaining, processes of living cells. Once built, the individual models serve as digital laboratories, allowing formerly labor-intensive tests to be conducted with the click of a mouse. Yizhak’s algorithm, which she calls a “metabolic transformation algorithm,” or MTA, can take information about any two metabolic states and predict the environmental or genetic changes required to go from one state to the other.
“Gene expression” is the measurement of the expression level of individual genes in a cell, and genes can be “turned off” in various ways to prevent them from being expressed in the cell. In the study, Yizhak applied MTA to the genetics of aging. After using her custom-designed MTA to confirm previous laboratory findings, she used it to predict genes that can be turned off to make the gene expression of old yeast look like that of young yeast. Yeast is the most widely used genetic model because much of its DNA is preserved in humans.
Some of the genes that the MTA identified were already known to extend the lifespan of yeast when turned off. Of the other genes she found, Yizhak sent seven to be tested at a Bar-Ilan University laboratory. Researchers there found that turning off two of the genes, GRE3 and ADH2, in actual, non-digital yeast significantly extends the yeast’s lifespan.
“You would expect about three percent of yeast’s genes to be lifespan-extending,” said Yizhak. “So achieving a 10-fold increase over this expected frequency, as we did, is very encouraging.”
Hope for humans
Since MTA provides a systemic view of cell metabolism, it can also shed light on how the genes it identifies contribute to changes in genetic expression. In the case of GRE3 and ADH2, MTA showed that turning off the genes increased oxidative stress levels in yeast, thus possibly inducing a mild stress similar to that produced by calorie restriction.
As a final test, Yizhak applied MTA to human metabolic information. MTA was able to identify a set of genes that can transform 40-to-70 percent of the differences between the old and young information from four different studies. While currently there is no way to verify the results in humans, many of these genes are known to extend lifespan in yeast, worms, and mice.
Next, Yizhak will study whether turning off the genes predicted by MTA prolongs the lifespan of genetically engineered mice. One day, drugs could be developed to target genes in humans, potentially allowing us to live longer. MTA could also be applied to finding drug targets for disorders where metabolism plays a role, including obesity, diabetes, neurodegenerative disorders, and cancer.
——————————————————————————–
For more genetics research news from Tel Aviv University, click here.
Keep up with the latest AFTAU news on Twitter: http://www.twitter.com/AFTAUnews.
Pingback: Digital health – health and fitness wearables, Part 3: Heart Rate Variability: Principles and Science and Practical Measuring Devices | AGING SCIENCES – Anti-Aging Firewalls