
By James P Watson with assistance and added commentary by Vince Giuliano
This is a story of how we got here to our obesity and metabolic disease epidemic. Goes a bit back – around 30-40 million years – with another important Chapter that started about 45 years ago. It is a story about climate change, what happened during global cooling, about industrialized agriculture, and about dietary changes wayback then and now. And about how the gooey contents of thousands of grimy black rail tank cars seen everywhere in Iowa and the Midwest is making us sick because of what happened millions of years ago in icy Europe.
Once we were Great Apes
 Image source Eocene Primates – Library of Congress vintage illustration
Image source Eocene Primates – Library of Congress vintage illustration
We still are Great Apes in fact, though we don’t remember most things we have gone through in our long and challenging primate past. This story starts with two specific loss-of-function gene mutations that occurred long ago during our primate evolution, mutations that have huge implications today for dietary choices. These two gene mutations play a major causal role in the current world-wide epidemic of obesity, metabolic syndrome (central truncal obesity, hypertriglyceridemia, low HDL, high BO, and elevated FBS), type 2 diabetes, cardiovascular disease, and accelerated aging.
No genotyping are needed for these two gene mutations that are mostly “primate specific.” These two mutations occurred during the late Eocene and mid-Miocene periods of primate evolution, during periods of global cooling and accelerated primate evolution. Together, the two gene mutations mean that we are all “double knock-outs” for very important genes that were essential for our survival as primate predecessors. The problem now, however, is that these knock-outs lead to high fructose-induced visceral fat build-up, which is the cause of metabolic syndrome, obesity, and type 2 diabetes (the 3 major disease scourges of our times).
The basic argument is pretty simple. It is that during these ancient epochs of global cooling, our simian ancestors underwent two forever loss-of function gene mutations, one mutation (30-40 million years ago) wiping out our ability to synthesize Vitamin C, the other mutation (10-15 million years ago) undermining our ability to degrade uric acid. Back then, these mutations together enabled us to use fructose to accumulate layers of fat which helped us to stay alive during the long cold winters, a time when you could not go to a trendy Fusion restaurant, or pick up healthy ingredients from a Whole Foods, or order a pizza from a local shop. Nowadays fructose seems to be everywhere. High fructose corn syrup is stuff carried in grimy black railroad tank cars and is universally included in thousands of manufactured food and drink products. And the same layers of visceral fat give us the not-so-nice things we mentioned earlier – the current world-wide epidemic of obesity, metabolic syndrome (central truncal obesity, hypertriglyceridemia, low HDL, high BO, and elevated FBS), type 2 diabetes, cardiovascular disease, and accelerated aging.
So, those two gene mutations – 50 and 15 million years ago – together with universal use of high fructose corn syrup in manufactured foods – starting about 45 years ago – have combined to give us a major health epidemic.
The good news is that there are some very specific dietary and lifestyle interventions that can successfully prevent 100% of this modern day, 3-disease scourge. However, to really understand why these specific dietary changes makes such a difference in metabolic syndrome, obesity, and T2D, it is crucial to understand these two gene mutations. These gene mutations are the “crux” of the modern day scourge – visceral fat. To understand how we reached this condition, we can start by considering climate change that occurred during primate evolution – the global cooling periods of the Eocene and Miocene epochs.
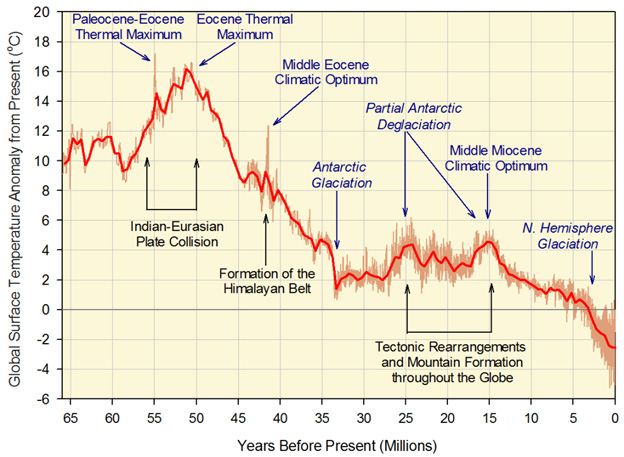
Image source Periods of global cooling
Humans are “Double Knock-outs”
The GLO gene loss – 30-40 million years ago
“THE BEST IDEA IS TO GET FAT USING FRUCTOSE”
Humans lost the ability to synthesize Vitamin C due to the deletion of multiple Exons in the L-gulono lactone oxidase (GLO) gene. In non-primate mammals, Vitamin C may be the single most important non-enzyme antioxidant. Denham Harmon’s “Free Radical Theory if Aging” would predict that a mutation in such a key antioxidant gene would not survive the “survival of the fittest” world of evolution. Denham Harmon’s theory would predict all animals with this mutation would become extinct. Instead, this mutation has survived 40 million years along the “evolutionary road kill highway”. Today, all primates carry this mutation except for prosimians, a feat that defies Hardy-Weinberg equilibriums in gene alleles. Today the GLO gene is so damaged that it is called a pseudogene and has not been transcribed to create a protein in 30 million years! The old explanations for why all primates have this loss-of-function mutation (except prosimians) does not “hold water”. For instance, Linus Pauling suggested that this loss-of-function mutation occurred because our ancestors were eating fruit and getting enough Vitamin C this way. But this does not explain why the mutation has reached nearly 100% penetrance! For such widespread expression of a mutant gene, there has to be what the evolutionary biologists call “positive selection pressure”. The abundance of fruit is not a “selection pressure”, especially in cold weather when there were no grocery stores open in the winter that carried fruit in the primate food section, 30 million years ago! LOL!
The major advantage that GLO knock-out primates may have had was their ability to accumulate fat. Fat may have helped these GLO gene mutant primates survive cold weather. Low Vitamin C levels trigger fat build-up by at least two molecular mechanisms:
- Low Vit C reduces Uric acid secretion (Uric acid increases NADPH oxidase activity inside the cell, triggering oxidative stress and insulin resistance)
- Vit C partially blocks the effects of fructose-mediated induction of metabolic syndrome. Low Vit C levels do the opposite – promote fructose-mediated metabolic syndrome
In the late Eocene period, when the GLO gene mutation flourished, global cooling occurred due to declines in atmospheric CO2. This resulted in cold winters and seasonal food shortages. Richard Johnson and colleagues at the U of Colorado have hypothesized that the primates carrying the GLO gene knock-out were able to build up fat stores better than the primates carrying functional copies of the GLO gene. This is a much more plausible explanation for why this knock-out allele reaches nearly 100% penetrance in Eocene primates.
The Urate Oxidase (uricase) gene loss – a multistage loss at 15 million and 9.8 million years ago
“BETTER YET, USE URIC ACID TO HELP YOU GET FAT CONSUMING FRUCTOSE“
Almost all primates carry another loss-of-function mutation in another antioxidant gene.
Again, Denham Harmon’s “Free radical theory of aging” would predict that any mammal carrying such a gene mutation would become extinct! Instead, the knock-outs survived and the functional gene primates died off! (Sorry Mr. Harmon, but you should have studied evolution before you made up your crazy theory!) This 2nd anti-oxidant gene knock-out occurred much more recently – 10-15 million years ago, when another period of global cooling occurred. This time another ice age occurred during the Miocene period of evolution. From comparative genetic studies of various primates, It appears that mutations started showing up in the promoter region of the urate oxidase gene. This effectively down-regulated expression of the gene long before it became nonfunctional. Then multiple mutations started occurring in the protein coding portions of the urate oxidase gene (I.e. in the 2nd and 5th exons). Gibbons developed a nonsense mutation in codon 33 and a 13-bp deletion in the ORF. Chimps developed 3 deleterious mutations. In great apes, the urate oxidase gene mutations occurred approximately 15 million years ago and in lesser apes, the uricase gene mutations occurred 9.8 million years ago. Since humans are genetically more similar to the great apes, we share the same mutation in codon 33.
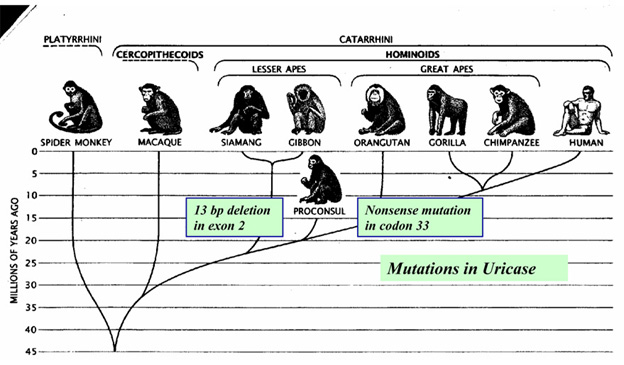
It is thought that this series of uricase mutations primarily happened in Europe where the winters were long and cold and fresh fruit nonexistent. Since there has always been year-round fruit in parts of Africa, it seems doubtful that the mutations happened only there. Later, according to the Back to Africa hypothesis, hominoid migrations back to Africa resulted in the mutations being carried back there.
Outside of the cell uric acid is considered an anti -oxidant, but inside the cell, there is evidence that uric acid promotes oxidative stress by multiple mechanisms. First of all, uric acid causes mitochondrial dysfunction, increasing the efflux of mitochondrial free radicals. Uric acid also activates NADPH oxidase, which is a plasma membrane bound producer of free radicals. Thus uric acid causing oxidative stress via mitochondrial and plasma membrane sources of free radicals. Why would a loss-of-function gene mutation like this survive if it increased oxidative stress? Again, the old theories fail to explain the ubiquitous loss of urate oxidase gene function in the large majority of primates. Unlike the GLO gene mutations (which all appear to come from a very old common ancestor), many different urate oxidase gene mutations occurred independently, suggesting again that some natural “selection pressure” existed between 9.8 and 15 million years ago. What was it? Again, cold climates encroached on primate habitats, giving mutants with fat-gaining abilities a survival advantage over their lean counterparts. As it turns out, uric acid is actually a great molecule to promote fat build-up, by increasing the absorption of fructose in the intestine and the liver. Uric acid increases gene expression of Glut5 and Glut2, which are the two major transporters if fuctose.
So, the metabolism of fructose causes much oxidative stress in cells (compared to glucose), a quite “good thing” 10-15 million years ago. Body fat accumulation gave us a distinct survival advantage back then when global cooling was occurring for the 2nd time. More body fat in periods of food scarcity may not be the only advantages conveyed by these mutations. “We suggest that both mutations may have provided a survival advantage to early primates by helping maintain blood pressure during periods of dietary change (including low salt) and environmental stress(ref) (ref).”
Enter the age of HFCS (last 45 years) –
“WHOOPS GETTING FAT USING FRUCTOSE IS NOW TURNING OUT TO BE A VERY BAD IDEA“
The sugar fructose is at the epicenter of the current perfect storm of the metabolic disease epidemics we are experienced today. First, because of the two ancient gene mutations described above which make our metabolisms extremely susceptible to wreckage by fructose, and second because of the massive industrial scale usage of high fructose corn syrup (HFCS) as the preferred sweetner in practically all manufactured food and drink products. As an industrial food product, HFCS is ideal in several respects. First of all, it is extremely cheap because of the industrial scale of corn production subsidized by tens of billions of dollars of federal subsidies. It is produced, shipped and used on mega-scales. A single train of HFCS cars like that in the video here can ship a million gallons of it. It keeps well and blends into canned soups, packaged meals, cookies, cakes, breads, ice cream, canned fruits, salad dressings, breakfast cereals, sweetened yogurts, cheese products, cough drops, jams, jellys, candy bars, snack packs, ketchup and barbecue sauce, coffee creamers, – you name it. Thousands or tens of thousands of food products, including “nutrition bars” and “natural juice cocktails.” Adding HFCS can delay food product expiration dates and must people think fructose is from fruit and therefore must be good. And it is used in gigantic CAFOs — Concentrated Animal Feeding Operations.

This Youtube video of a corn syrup train gives an idea of the scale of high fructose corn syrup in our society:
“High fructose corn syrup” used to evoke an image in me (Vince) of a wholesome and good product, made out of fresh juice squeezed by hand from healthy corn kernels by Aztec maidens. Perhaps, a cousin of strawberry or orange juice, definitely related to real fruit.
 A corn maiden image
A corn maiden image
Very nice, but far from what is. HFCS is not chemically the same as anything found in nature and is manufactured in large industrial facilities that look a lot like oil refineries. The process is complicated, involving large vats of murky fermenting liquid with Aspergillus fungus, liquid chromatography and multiple steps of chemical tweaking.

HFCS manufacturing plant in Cedar Rapids Iowa Image source, one of 14+ such plants in the corn belt.
Food and drink products with HFCS are sweeter and cheaper than products made with cane sugar. Manufacturers of soft drinks have been able to increase bottle size dramatically without charging much more – from 8 to 20 ounces. And machines that inexpensively dispense immense soda drinks are in all fast food restaurants, pizza shops and convenience stores. While there has been little visable added price cost to consumer, the true costs have been massive, to the consumer and to our health system. The costs show up as increased obesity, metabolic syndrome, diabetes, and chronic disease. So, today, fructose intake has sky rocketed, with the annual consumption of sugar increasing from 4 lbs/year to 130-200 lbs per year from 1700 AD to 2015 AD. Fructose (compared to glucose) is a major cause of metabolic syndrome, obesity, and type 2 diabetes. And tooth decay.
If humans did not carry these two loss of function gene mutations, the high fructose corn syrup and high sugar intake that characterizes the Western Diet might not have produced the current epidemic of obesity, metabolic syndrome, and type 2 diabetes. However with this 30-50 fold increase in total sugar and an even greater increase in fructose, our “double knock-out genotype” that protected us from cold winters and famine now has become a true Genetic Scourge.
The health news in the last decade has not been kind to HFCS. HFCS can promote abnormal heart growth, increasing the risk for heart failure. According to a 2015 study(ref). HFCS has been linked to opioid addiction (ref). Further, one study in 2015 concluded that “Nearly half of all commercial high-fructose corn syrup (HFCS) used in processed foods like soda pop, ketchup and candy is tainted with toxic mercury, according to a peer-reviewed study published in the journal Environmental Health. Among 20 samples of commercial HFCS tested for the heavy metal, nine tested positive, says the Institute for Agriculture and Trade Policy (IATP), the consumer group that spearheaded the study. — According to The Washington Post (WP), the average American consumes roughly 12 tablespoons of HFCS daily, while teenagers and other “high consumers” are believed to consume up to 80 percent more, or roughly 22 tablespoons, of the substance every single day. Based on the findings of the study, this means that mercury is being ingested at levels never before seen, presenting serious health risks that could cause permanent health damage.”
HFCS seems mainly to be a US and Canadian problem. Europe countries like UK don’t have this problem because they do not authorize the use of this ingredient. It is banned in the UK by a production quota. In Canada, favorable treatment of HFCS under NAFTA rules may have contributed significantly to obesity. “The study, covering 1985-2000, found that lower tariffs on HFCS were associated with an increase of about 41.6 kcal in caloric sweeteners supplied and likely consumed per person per day in Canada. This increase in the supply of HFCS correlates with a large rise in obesity rates, from 5.6% in 1985 to 14.8% in 1998, as well as increases in diabetes. Even seemingly small increases in caloric intake can contribute to weight gain, with small daily increases adding up over time(ref).”
The age of HFCS may be topping off as we write this. From Wikipedia: “In August 2016 in a move to please consumers with health concerns, McDonald’s announced they would be replacing all HFCS in their buns with sucrose (table sugar) and would cut out preservatives and other artificial additives from their menu items.[73]Marion Gross, senior vice president of McDonald’s stated, “We know that they [consumers] don’t feel good about high-fructose corn syrup so we’re giving them what they’re looking for instead.”[73] Over the early 21st century, other companies such as Yoplait, Gatorade, and Hershey’s also phased out HFCS, replacing it with conventional sugar because consumers perceived sugar to be ‘healthier’.[74][75] Companies such as PepsiCo and Heinz have also released products that use sugar in lieu of HFCS, although they still sell HFCS-sweetened products.[71][74] ”
If you are concerned with deleterious gene mutations, start (and possibly end) here
So, if you want to do genotyping to determine a personalized dietary plan, I suggest gene sequencing your GLO gene, located on the short arm of chromosome 8; and gene sequencing your urate oxidase gene, located on chromosome 1. I can tell you now what you will find – these genes don’t work anymore! What does this mean? It means you are prone to Western Diet-induced obesity, metabolic syndrome, hypertriglyceridemia, low HDL, High BP, beer belly, coronary artery disease, Stroke, and type II diabetes!
These two gene mutations are 1000 times more important than all of those SNPs that the nutrigenomics companies are trying to peddle on ignorant customers! First and foremost, accept your identity as a “double knock-out”! Your ancestors have carried these gene mutations for millions of years! Don’t sweat the single point mutations that don’t even come close to the significance of the GLO and uricase mutations! And there is a “primate specific diet” that you should be on if you genotype your primate genes.
What you can do with diet
The “personalized primate diet” that you should be on as a result of your genetic analysis should include the following:
- Exogenous Vitamin C supplementation. This of course is what many of us do. Unfortunately this may not work very well because of the poor GI absorption of Vit C. Gene therapy with a messenger RNA encoding for the GLO mRNA would probably be a better way to go, since many mammals endogenously synthesize up to 14 grams of Vit C per day. Another way to go could be to take liposomal Vitamin C.
- Avoid “added fructose” like a plague! Fructose is a major cause of hyperuricemia, via a mechanism described in Richard Johnson’s paper below. The mechanism involves transient ATP depletion, and AMP deamination by AMO .deaminase, which supplies more purines to the Xanthine/Uric acid pathway. Avoid manufactured foods, carry a magnifying glass and read the labels in supermarkets and don’t buy anything with high fructose corn syrup listed as an ingredient. Fructose naturally present in fresh fruits and vegetables is not a problem.
- Reduce animal protein and increase plant-derived proteins. Animal proteins have a lot more hypoxanthine and xanthine in the protein, increasing uric acid
- Reduce alcohol drinks that tend to promote a “beer belly.” In general, this means “beer” (that is where beer bellies come from). The discussion part of Richard Johnson’s paper explains how beer increases uric acid.
- Consider taking allopurinol, a xanthine oxidase inhibitor medication used specifically to control uric acid levels. It only costs pennies a day and I would recommend a goal of achieving uric acid levels that mirror those found in mammals without the uricase mutation.
References:
Theodore E. Woodward Award: The Evolution of Obesity: Insights from the Mid- Miocene 2015
“All humans are double knockouts. Humans lack the ability to synthesize vitamin C due to a mutation in L-gulono-lactone oxidase that occurred during the late Eocene, and humans have higher serum uric acid levels due to a mutation in uricase that occurred in the mid Miocene. In this paper we review the hypothesis that these mutations have in common the induction of oxidative stress that may have had prosurvival effects to enhance the effects of fructose to increase fat stores. Fructose was the primary nutrient in fruit which was the main staple of early primates, but this food likely became less available during the global cooling that occurred at the time of these mutations. However, in today’s society, the intake of fructose, primarily in the form of added sugars, has skyrocketed, while the intake of natural fruits high in vitamin C has fallen. We suggest that it is the interaction of these genetic changes with diet that is responsible for the obesity epidemic today. Hence, we propose that Neel’s thrifty gene hypothesis is supported by these new insights into the mechanisms regulating fructose metabolism.”
Added Fructose 2015
“Data from animal experiments and human studies implicate added sugars (eg, sucrose and high-fructose corn syrup) in the development of diabetes mellitus and related metabolic derangements that raise cardiovascular (CV) risk. Added fructose in particular (eg, as a constituent of added sucrose or as the main component of high-fructose sweeteners) may pose the greatest problem for incident diabetes, diabetes-related metabolic abnormalities, and CV risk. Conversely, whole foods that contain fructose (eg, fruits and vegetables) pose no problem for health and are likely protective against diabetes and adverse CV outcomes. Several dietary guidelines appropriately recommend consuming whole foods over foods with added sugars, but some (eg, recommendations from the American Diabetes Association) do not recommend restricting fructose-containing added sugars to any specific level. Other guidelines (such as from the Institute of Medicine) allow up to 25% of calories as fructose-containing added sugars. Intake of added fructose at such high levels would undoubtedly worsen rates of diabetes and its complications. There is no need for added fructose or any added sugars in the diet; reducing intake to 5% of total calories (the level now suggested by the World Health Organization) has been shown to improve glucose tolerance in humans and decrease the prevalence of diabetes and the metabolic derangements that often precede and accompany it. Reducing the intake of added sugars could translate to reduced diabetes-related morbidity and premature mortality for populations.”
“Reduced AMP kinase (AMPK) activity has been shown to play a key deleterious role in increased hepatic gluconeogenesis in diabetes, but the mechanism whereby this occurs remains unclear. In this article, we document that another AMP-dependent enzyme, AMP deaminase (AMPD) is activated in the liver of diabetic mice, which parallels with a significant reduction in AMPK activity and a significant increase in intracellular glucose accumulation in human HepG2 cells. AMPD activation is induced by a reduction in intracellular phosphate levels, which is characteristic of insulin resistance and diabetic states. Increased gluconeogenesis is mediated by reduced TORC2 phosphorylation at Ser171 by AMPK in these cells, as well as by the up-regulation of the rate-limiting enzymes PEPCK and G6Pc. The mechanism whereby AMPD controls AMPK activation depends on the production of a specific AMP downstream metabolite through AMPD, uric acid. In this regard, humans have higher uric acid levels than most mammals due to a mutation in uricase, the enzyme involved in uric acid degradation in most mammals, that developed during a period of famine in Europe 1.5 × 107 yr ago. Here, working with resurrected ancestral uricases obtained from early hominids, we show that their expression on HepG2 cells is enough to blunt gluconeogenesis in parallel with an up-regulation of AMPK activity. These studies identify a key role AMPD and uric acid in mediating hepatic gluconeogenesis in the diabetic state, via a mechanism involving AMPK down-regulation and overexpression of PEPCK and G6Pc. The uricase mutation in the Miocene likely provided a survival advantage to help maintain glucose levels under conditions of near starvation, but today likely has a role in the pathogenesis of diabetes.—Cicerchi, C., Li, N., Kratzer, J., Garcia, G., Roncal-Jimenez, C. A., Tanabe, K., Hunter, B., Rivard, C. J., Sautin, Y. Y., Gaucher, E. A., Johnson, R. J., Lanaspa, M. A. Uric acid-dependent inhibition of AMP kinase induces hepatic glucose production in diabetes and starvation: Evolutionary implications of the uricase loss in hominids.”
“All human beings are uricase knockouts; we lost the uricase gene as a result of a mutation that occurred in the mid-Miocene epoch approximately 15 million years ago. The consequence of being a uricase knockout is that we have higher serum uric acid levels that are less regulatable and can be readily influenced by diet. This increases our risk for gout and kidney stones, but there is also increasing evidence that uric acid increases our risk for hypertension, kidney disease, obesity, and diabetes. This raises the question of why this mutation occurred. In this article we review current hypotheses. We suggest that uric acid is a danger and survival signal carried over from the RNA world. The mutation of uricase that occurred during the food shortage and global cooling that occurred in the Miocene epoch resulted in a survival advantage for early primates, particularly in Europe. Today, the loss of uricase functions as a thrifty gene, increasing our risk for obesity and cardiorenal disease.”
Redefining metabolic syndrome as a fat storage condition based on studies of comparative physiology 2013
“Objective: The metabolic syndrome refers to a constellation of signs including abdominal obesity, elevated serum triglycerides, low HDL-cholesterol, elevated blood pressure, and insulin resistance. Today approximately one third of the adult population has the metabolic syndrome. While there is little doubt that the signs constituting the metabolic syndrome frequently cluster, much controversy exists over the definition, pathogenesis, or clinical utility. Design and Methods: Here we present evidence from the field of comparative physiology that the metabolic syndrome is similar to the biological process that animals engage to store fat in preparation for periods of food shortage.”
Fructose, uricase, and the Back-to-Africa hypothesis 2010
“In the Middle Miocene (approximately 17 to 12 Ma) at least two radiations of fossil apes from East Africa into Eurasia occurred, and, while controversial, some paleoanthropological studies suggest that one of the Eurasian lineages may have returned to Africa to evolve into humans and the African apes. Here, we present a novel argument supporting this hypothesis. Specifically, the global cooling that occurred in the middle Miocene rendered hominoids living in Europe at risk for starvation as seasonal climate change resulted in less availability of fruits during the winter months. During this time, a mutation in uricase occurred in early hominids that resulted in a rise in serum uric acid. Uric acid has been found to potentiate the effect of fructose to increase fat stores, suggesting that the mutation provided a survival advantage. Such a survival advantage would have been less likely to occur in Africa, where the continued presence of tropical rainforests would have been more likely to provide food throughout the year. Furthermore, Miocene apes in Europe were in protected sites where geographic isolation could have allowed the uricase mutation to be rapidly expressed in the entire population. While speculative, we suggest that the uricase mutation supports an extra-Africa origin of humans.”
“Humans have relatively low plasma ascorbate levels and high serum uric acid levels compared to most mammals due to the presence of genetic mutations in L-gulonolactone oxidase and uricase, respectively. We review the major hypotheses for why these mutations may have occurred. In particular, we suggest that both mutations may have provided a survival advantage to early primates by helping maintain blood pressure during periods of dietary change and environmental stress. We further propose that these mutations have the inadvertent disadvantage of increasing our risk for hypertension and cardiovascular disease in today’s society characterized by Western diet and increasing physical inactivity. Finally, we suggest that a “planetary biology” approach in which genetic changes are analyzed in relation to their biologic action and historical context may provide the ideal approach towards understanding the biology of the past, present and future.”
Humans have relatively low plasma ascorbate levels and high serum uric acid levels compared to most mammals due to the presence of genetic mutations in L-gulonolactone oxidase and uricase, respectively. We review the major hypotheses for why these mutations may have occurred. In particular, we suggest that both mutations may have provided a survival advantage to early primates by helping maintain blood pressure during periods of dietary change and environmental stress. We further propose that these mutations have the inadvertent disadvantage of increasing our risk for hypertension and cardiovascular disease in today’s society characterized by Western diet and increasing physical inactivity. Finally, we suggest that a “planetary biology” approach in which genetic changes are analyzed in relation to their biologic action and historical context may provide the ideal approach towards understanding the biology of the past, present and future.
Uric acid, evolution and primitive cultures 2004 “Hypertension is epidemic and currently affects 25% of the world’s population and is a major cause of stroke, congestive heart failure, and end-stage renal disease. Interestingly, there is evidence that the increased frequency of hypertension is a recent event in human history and correlates with dietary changes associated with Westernization. In this article, we review the evidence that links uric acid to the cause and epidemiology of hypertension. Specifically, we review the evidence that the mutation of uricase that occurred in the Miocene that resulted in a higher serum uric acid in humans compared with most other mammals may have occurred as a means to increase blood pressure in early hominoids in response to a low-sodium and low-purine diet. We then review the evidence that the epidemic of hypertension that evolved with Westernization was associated with an increase in the intake of red meat with a marked increase in serum uric acid levels. Indeed, gout and hyperuricemia should be considered a part of the obesity, type 2 diabetes, and hypertension epidemic that is occurring worldwide. Although other mechanisms certainly contribute to the pathogenesis of hypertension, the possibility that serum uric acid level may have a major role is suggested by these studies.”
“Uric acid has historically been viewed as a purine metabolic waste product excreted by the kidney and gut that is relatively unimportant other than its penchant to crystallize in joints to cause the disease gout. In recent years, however, there has been the realization that uric acid is not biologically inert but may have a wide range of actions, including being both a pro- and anti-oxidant, a neurostimulant, and an inducer of inflammation and activator of the innate immune response. In this paper, we present the hypothesis that uric acid has a key role in the foraging response associated with starvation and fasting. We further suggest that there is a complex interplay between fructose, uric acid and vitamin C, with fructose and uric acid stimulating the foraging response and vitamin C countering this response. Finally, we suggest that the mutations in ascorbate synthesis and uricase that characterized early primate evolution were likely in response to the need to stimulate the foraging “survival” response and might have inadvertently had a role in accelerating the development of bipedal locomotion and intellectual development. Unfortunately, due to marked changes in the diet, resulting in dramatic increases in fructose- and purine-rich foods, these identical genotypic changes may be largely responsible for the epidemic of obesity, diabetes and cardiovascular disease in today’s society.”
Soluble Uric Acid Activates the NLRP3 Inflammasome 2017
“Uric acid is a damage-associated molecular pattern (DAMP), released from ischemic tissues and dying cells which, when crystalized, is able to activate the NLRP3 inflammasome. Soluble uric acid (sUA) is found in high concentrations in the serum of great apes, and even higher in some diseases, before the appearance of crystals. In the present study, we sought to investigate whether uric acid, in the soluble form, could also activate the NLRP3 inflammasome and induce the production of IL-1β. We monitored ROS, mitochondrial area and respiratory parameters from macrophages following sUA stimulus. We observed that sUA is released in a hypoxic environment and is able to induce IL-1β release. This process is followed by production of mitochondrial ROS, ASC speck formation and caspase-1 activation. Nlrp3−/−macrophages presented a protected redox state, increased maximum and reserve oxygen consumption ratio (OCR) and higher VDAC protein levels when compared to WT and Myd88−/− cells. Using a disease model characterized by increased sUA levels, we observed a correlation between sUA, inflammasome activation and fibrosis. These findings suggest sUA activates the NLRP3 inflammasome. We propose that future therapeutic strategies for renal fibrosis should include strategies that block sUA or inhibit its recognition by phagocytes. — Uric acid, the product of purine catabolism, is a damage-associated molecular pattern (DAMP) released from ischemic tissues and dying cells1,2. Once crystalized, uric acid activates the immune system3,4, since it acts as a pro-oxidant molecule that reduces nitric oxide availability1, increases the production of reactive oxygen species (ROS), stimulates chemotaxis and also activates NF-κB and MAPK pathways1. Uric acid crystals also induce the release of proinflammatory cytokines such as IL-1β4,5, a signaling molecule secreted when the formation of a high molecular weight complex named “inflammasome” is activated6. — Crystals trigger inflammasome activation4,7,8,9 mainly through frustrated phagocytosis, a process characterized either by aberrant actin polymerization10 or by lysosomal damage11, in which lysosome contents leak into the cytosol. Lysosomal proteases, once in the cytosol, in addition to acting as DAMPs12, are able to digest vital proteins and affect other organelles, such as the mitochondria. In addition, uric acid crystals can directly engage cellular membranes without the involvement of any known cellular receptor. Recent evidences demonstrate that serum uric acid levels are also associated with inflammatory effects, such as in gout-related disease and preeclampsia13,14. However, it is still unknown if soluble uric acid (sUA) activates the inflammasome complex and lead to IL-1β production. Primates lost the uricase gene and the direct consequence is that they have higher serum uric acid levels than other animals15. The mutation in the uricase gene that occurred during food scarcity and global cooling resulted in a survival advantage at that time16. Today, however, it is associated with hypertension, kidney disease, obesity and diabetes17. Accumulating evidence has revealed a positive relationship between serum uric acid levels and cardiovascular mortality in patients with chronic kidney disease18,19. Despite an unknown mechanism, local accumulation of uric acid may activate the inflammasome complex, leading to inflammation and fibrosis20.”
