
The Molecules that Play Dr. Jeckel and Mr. Hyde in Aging, Alzheimer’s Disease, and Type II Diabetes
By James P Watson, with summary and contributions from Vince Giuliano
Introduction – GSK-3s – The “Dr. Jeckel and Mr. Hyde” Molecules
Have you ever heard of Glycogen Synthase Kinase 3 alpha or beta? If you haven’t, don’t feel bad…..they do so much more than just inhibit glycogen synthesis. If you are interested in what causes aging or Alzheimer’s disease, however, you better learn about GSK-3, because this is big….really big! Why hasn’t GSK-3s received more press? Maybe it is because no PR agent has been hired to “pitch” this poorly named enzyme. So I am going to pretend I am a PR firm hired to develop an advertising campaign for GSK-3α and GSK-3β. The first thing that PR firms do is to develop a catchy “tag line” to describe the GSK-3s. One possibility would be “GSK-3: We control much more than Glycogen Synthesis” (That isn’t that catchy, but GSKs do control over 50 molecular pathways). Most people don’t even know what glycogen is, so maybe a better tag line would be “GSK-3: the Master Molecular Multi-tasker” (every modern woman can relate to that!). Another one that might get attention is the Great Satan Kinase, but actually that slights it’s good side. For the engineers or science geeks who may read this blog, we have another PR slogan for GSK-3: “The Multipurpose Signal Amplifier and Multipurpose Signal Dampener”. Even that technical mumbo-jumbo doesn’t really reflect how powerful the GSK-3 enzymes are, however. For this reason, our PR firm has finally decided to give GSK-3s a “human face” –
The Molecular Dr. Jeckel and Mr. Hyde! 
They are your worst enemy and your best friend. They can throw a “monkey wrench” in all of the major molecular signaling cascades (like the Insulin/IGF pathway, the mTOR pathway, etc.). They can keep you healthy or they can make you age. They can cause Alzheimer’s disease and Diabetes or they can protect you from these diseases. In fact, because the way they function as amplifiers, GSK-3s can kill your cells by activating apoptosis pathways. This is why they are both “good” and “bad” for you.
This is the first of what we expect to be a series of blog entries on less-familiar but very important and well-studied molecules encountered in humans which have major impacts on aging and health – the Strange But Powerful Molecules Series. GSK-3s have been studied in the cancer research community for over 30 years, but have largely been ignored in the aging research community despite their profound impacts on aging processes. Additional posts in the series will be concerned with other lesser-known candidates of similar great importance, starting with the P66shc protein and with the non-coding gene known as ANRIL. We characterize GSK-3 kinases, what they do, their impacts on biological pathways, why they are of critical importance for certain disease processes and aging, and about the ways they can be activated or turned off.
KEY POINTS
As is often the case, this blog gets quite complicated. So, we summarize some of the most important points here.
- The GSK-3s are important because they “signal amplifiers” that turn on genes or up regulate enzymes, but are also “signal dampeners” that turn off genes or down regulate enzymes. They have about 50 targets, approximately half of which are transcription factors and half are other (non-transcription factor) enzymes within the cell. They have so many different impacts on the cell that they are best referred to as “molecular multi-taskers”. They play important roles in multiple disease processes and in aging itself.
- “In the pathways in which GSK-3 acts, it serves as a key regulator of normal cellular functions. However, when GSK-3 is dysregulated, it also has a key role in causing diseases such as diabetes, Alzheimer’s disease, bipolar disorder and cancer(ref).” GSK-3s now are well documented to play key roles also in Chronic Traumatic Encephalopathy, Fronto-temporal dementia, Multiple Sclerosis, Diabetes type II, colitis, and arthritis. “Aberrant activity of GSK-3 has also been implicated in the pathologies of many diseases and disorders such as metabolic disorders (diabetes, atherosclerosis, heart disease), neurological disorders (Parkinson’s, Alzheimer’s, amyotrophic lateral sclerosis, schizophrenia, bipolar disorder, mood disorders), cancer and aging (cellular senescence, cancer stem cells, control of stem cell pluripotency and differentiation), immune disorders and other maladies among others(ref).3, 4, 5, 6, 7
- The GSK-3s exercise regulatory functions which impact on cell cycle and division dynamics and a DNA repair pathway.
- GSK’s directly impact aging since they can function as molecular mediators of the Insulin/IGF pathway and the mTOR pathway. However, when GSK-3s are inhibited by compounds such as lithium, they can “bypass” the normal methods of blocking these “pro-aging pathways” and can therefore make a direct impact towards reducing aging by inhibiting the Insulin/IGF pathway and inhibiting the mTOR pathway. This mechanism may be why GSK-3 inhibitors are such powerful compounds.
- The GSK’s act by phosphorylating key molecules. They prefer molecules which are already phosphorlated, so they can be thought of as being phosphorylation amplifiers. They can increase phosphorylation, by a factor of 100 or 1,000. With this kind of amplifier muscle, they can activate or shut down important signaling cascades.
- In normal resting tissues GSK-3s are active, turned on, Many kinds of signals can turn GSK-3s off, for examples, insulin signaling and growth factor signaling. The molecular process of turning GSK-3s off and degrading them is also phosphorylation, this time of the GSK-3 itself..
- GSK-3 plays important roles in both the innate and adaptive immune systems. For this reason, GSK-3 inhibition may help treat some types of autoimmune diseases where there is a hyperactive innate or adaptive immune system.
- GSK-3 plays a major role in inflammatory processes and in neuroinflammatory diseases in particular. Among its functions are control of various triggers of inflammation. And the production of pro and anti-inflammatory cytokines.
- There are two major isoforms of GSK-3, the alpha isoform which regulates longevity-related processes and he beta isoform which regulates aging and “shortivity”-related processes. The main molecular bodies of the two isoforms are very similar, but they have different molecular “tails.” What they do biologically is quite different.
- GSK-3 alpha when active negatively regulates at least three pro-aging pathways: mTOR, Wnt and P53. Basically, in normal quiescent cells GSK-3α restrains runaway aging due to activation of these pathways. If GSK-3α is not functioning normally, accelerated aging can occur.
- GSK-3β, on the other hand can promote the formation of both beta-amyloid plaques in the brain and tau tangles in nerve cells, the two main “usual suspect” causes of Alzheimer’s disease. Unlike GSK-3α, its expression increases with aging. It is thought to be an important long-term contributor to age-related neuro-degeneration. GSK-3β plays a role in signaling pathways like those involved in glycogen metabolism. It appears that its aberrant inhibition may be relevant in diabetes and neurodegenerative disorders. This isoform also promotes degradation of NF-kappaB and reduction of inflammatory conditions. So it is far from being all-bad.
- Many health-beneficial effects are associated with inhibition of GSK-3β and activation of the beta-catenin pathway, including bone mass maintenance, less osteoporosis, reduced insulin signaling, less atherosclerosis and lowered blood pressure. These might be limited however by competition for beta-catenin by pathways that respond to high states of oxidative stress, where beta-catenin is required as a cofactor. Therefore, effective therapeutic inhibition of GSK-3β might require additional anti-oxidant interventions such as suppression of P66shc.
- Blocking GSK-3β enhances expression of many proteins, an example being Nrf2 since GSK-3β phosphorylates Nrf2. This fact alone could explain some of the health benefits induced by lithium and a number of phytosubstances.
- However, since GSK-3β also phosphorylates the inflammatory factor BF-kappaB, it appears that in the presence of inflammation there may be a cost trade-off to blocking expression of GSK3β.
- There appears to be a number of effective GSK-3β inhibitors. Lithium is a good one, and many its neurological impacts and others of its health-producing effects could be due to GSK-3 inhibition, lower cholesterol, less oxidized LDL signaling.
- While beneficial effects can sometimes be realized by inhibiting GSK-3, care must be taken that such inhibition or activation does not also produce a negative effect via the other isoform or via a different pathway than that targeted. As a phosphorylation amplifier, like a PA system amplifier GSK-3t does not necessarily pay attention to the message itself.
THE SCIENCE OF GSKS
1. INTRODUCTION TO THE GSKs – serine/threonine protein kinases
They are “Molecular Multi-taskers” that functions as “signal amplifiers” for kinase cascades and transcription factor activation of genes
Discovered in the 1980s,GSKs3s were at first thought to merely be a “negative regulator” of glycogen synthase, and thus got their name. However, as more and more research has been done, the consensus emerged that GSKs exerts a broad, regulatory influence on 50+ targets and that glycogen synthesis inhibition was not the primary function of GSKs. Today we know that at least 25 transcription factors are activated/inhibited by GSK-3s and an additional 25+ other proteins are activated/inhibited by GSK-3s. For this reason, GSK-3s are are now viewed as “multitasking molecules” that act as “signal amplifiers” for many molecular pathways involving inflammation, gene expression, cell mobility, and apoptosis. Most recently, it has become clear that GSK-3s regulate lifespan as well. GSK-3s now are well documented to play a role in Alzheimer’s disease, Chronic Traumatic Encephalopathy, Fronto-temporal dementia, Multiple Sclerosis, Diabetes type II, Cancer, colitis, and arthritis.
GSK-3 do their “work” by phosphorylating substrates. A kinase phosphorylates substrates (a molecule on which an enzyme acts) by transferring phosphate groups to them from high-energy donor molecules. This in turn can have a profound effect on the properties of the phosphorylated substrate. “The phosphorylation state of a molecule, whether it be a protein, lipid, or carbohydrate, can affect its activity, reactivity, and its ability to bind other molecules. Therefore, kinases are critical in metabolism, cell signalling, protein regulation, cellular transport, secretory processes, and countless other cellular pathways(ref).” And, in its special role of being a kinase amplifier, GSK-3 can be considered to be “the mother of all kinases.”
The GSKs have a rather special property in that they only phosphorylate substrate proteins that are already phosphorylated, and in this respect function essentially as phosphorylation amplifiers. The literature speaks of already-phosphorylated substrates as being “primed,” and the GSKs impact differently on primed and unprimed substrates.
2. Substrates phospherylated by GSK-3 These include the following:
A. Transcription factor phosphorylation – GSK-3s activate at least 13 transcription factors and inactivate 12 transcription factors by phosphorylation. This, of course, implies contributing to turning genes on and off. As mentioned, GSKs prefer phosphorylating these transcription factors if they are “primed”. That means that another kinase must phosphorylate the transcription factor first. Only then, GSK-3 will come along and further activate the transcription factor. In this sense, GSKs are “amplifiers” of another signaling kinase cascade. (sort of like how p66shc is a “ROS amplifier” for redox signaling).
B. Non-transcription factor proteins – GSK-3s activate another 10 non-transcription factor proteins and inactivate another 14 non-transcription factor proteins. For these proteins, the GSK-3s also prefer activating the “pre-primed” proteins that have already been phosphorylated on another residue by another kinase cascade. Thus GSK-3s also function as a “kinase cascade amplifier”, rather than an original stimulus for the pathway. Think, then, of GSK signaling being like mass-media signaling, in some cases indifferent to the messages being communicated.
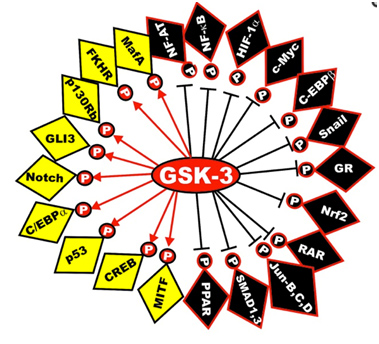
Here is a diagram of the transcription factors that are substrates of GSK phosphorylation. The yellow ones are activated and the black ones are inactivated. The diagram has left off the STAT transcription factors, which include STAT1, 2,5, and 6. The “big picture” here is that GSK-3s act as a “kinase signal amplifier” to increase gene expression or increase gene suppression by phosphorylating the transcription factor. GSK-3 inhibitors would have the opposite effect on these transcription factors, meaning that the black ones would be “yellow” and the yellow ones would be “black” with a GSK-3 inhibitor such as Lithium, a non-selective “ATP noncompetitive inhibitor” of GSK-3β, or maleimide derivatives, which are selective “ATP competitive inhibitors” of GSK-3β.
Reference: May 2014 GSK-3 as potential target for therapeutic intervention in cancer
Next is a diagram of the other proteins that are activated or inactivated by GSK-3s. The yellow ones are activated by GSK-3s, whereas the black ones are inactivated by GSK-3 phosphorylation. As mentioned below, a GSK-3 inhibitor such as Lithium, a Maleimide derivative, or a Thiadiazolidindione would exert the opposite effect, turning the yellow proteins “black” and the black proteins “yellow”.
Illustration reference: GSK-3 as potential target for therapeutic intervention in cancer
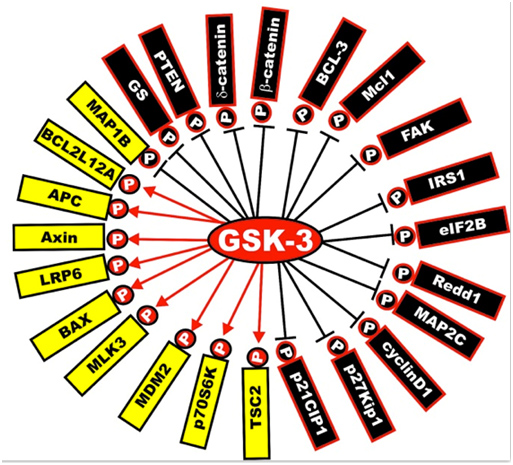
Frequent readers of this blog can note that GSK-3 impacts on a great many of the “usual suspects” related to disease processes and aging. That is their importance.
3. How GSK-3s are activated and how they are inactivated
Ser-9 or Ser-21phosphorylation inactivates GSK-3s.
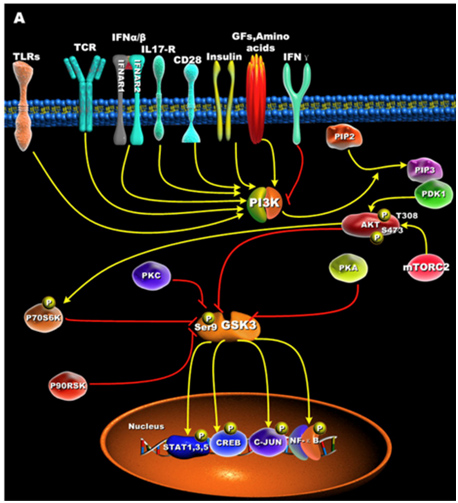
GSK-3s are normally activated when the cell is resting, but are inactivated by external stimuli. For instance, GSK-3s are actively inhibiting glycogen synthase until insulin signaling occurs. Then insulin inhibits GSK-3 and thus the “inhibition of an inhibitor of glycogen synthase” results in the synthesis of glycogen within the cell (liver, muscle, or fat cell). Most all of the ways that GSKs are inactivated is via phosphorylation of GSK-3. This “phosphoinactivation” pathway goes through PI3K. Growth factors, amino acids, Toll-like receptors (TLR), T cell receptor (TCR), CD28, and cytokine receptors have all been shown to mediate the PI3K/Akt-mediated “phosphor-inactivation” of GSK-3α and GSK-3β. GSKα is inactivated by the phosphorylation of Serine 21 (Ser-21). GSK-3β is inactivated by the phosphorylation of Serine 9 (Ser-9). Activation of Akt occurs when it is phosphorylated at thr-308 by PDK1 and with phosphorylation of Ser-473 by mTORC2.
Upon activation by PDK1 + mTORC2, Akt can phosphorylate GSK-3β at Ser-9. PKC, p70S6K, p90RSK, and PKA can phospho-inactivate GSK-3s as well. Inactivation of GSK-3s results in the activation of transcription factors important for regulating the innate and adaptive inflammatory response. The following diagram illustrates how GSK-3s are inactivated and what their downstream targets are:
Reference for illustration:2012 Glycogen Synthase Kinase 3: A Point of Convergence for the Host Inflammatory Response
4. Isoforms of GSK-3
There are two isoforms of GSK-3, alpha and beta.
a. The “alpha isoform” of GSK-3
– a longevity regulator – the mainly good guy
GSK-3α is encoded from a distinctly different gene than GSK-3β. GSK-3 isoforms also have different substrate preferences, especially in the brain (probably the same in peripheral tissues). GSK-3α expression does NOT increase with aging, whereas GSK-3β expression increases with aging. Whereas GSK-3β “knock out” embryos die by day 16 due to liver failure, GSK-3α knockout mice are viable and display increased insulin sensitivity and reduced fat mass. However, these GSK-3α “knock outs” still have metabolic and neurological abnormalities in adulthood.
GSK-3α is most abundantly expressed in the brain with particular abundance in the hippocampus, the neocortex, and the cerebellum. In the hippocampus, GSK-3β is more abundantly expressed than GSK-3α (GSK-3α is the “good GSK 3”, whereas GSK-3β is the “bad GSK-3,” though not always). There is mounting data that GSK-3α is an “upstream cause” of longevity and that GSK-3α negatively regulates three pro-aging pathways – mTOR, Wnt, and p53. However, GSK-3α does not seem to have that much of an effect on another pathway we would like to see suppressed in aging, the Insulin/IGF pathway (see below).
A. Inhibition of the mTOR pathway – GSK-3α inhibits mTOR and this is the primary mechanism by which GSK-3α exerts its “anti-aging effects”. In GSK-3α “knock out” mice, there is an increase in the phosphorylation of three mTORC1 targets: 4E-BP1, S6 Kinase, and ribosomal S6 protein. This results in a decline in autophagy of these GSK-3α “knock out” mice. The three markers of autophagy reduced in these GSK-3α “knock out” mice are ATG6, LC3-I/II ratio, and p62.
It also appears that GSK-3α is needed for autophagosome formation.
In summary, the “unrestrained” or “constitutive up regulation” of the mTOR pathway appeares to be the major mechanism by which GSK-3α “knock outs” exhibited accelerated aging.
B. Inhibition of Wnt signaling – The inhibition of Wnt signaling is a minor mechanism by which GSK-3α works. WNT signaling can be a “pro-aging pathway”. Klotho-deficient mice have accelerated aging due, in part, to increased Wnt signaling. GSK-3α inhibits Wnt signaling by phophosphorylating beta-catenin, which targets it for ubiquitin proteasomal degradation.
C. p53/sestrin pathway – Sestrins are target genes of p53. Sestrins protect cells from various stressors by functioning as antioxidants and as mTORC1 inhibitors. GSK-3β can regulate p53 activity, but I am not sure if GSK-3α can do the same.
D. Insulin/IGF-1 pathway – Although GSK 3α “knock out” mice have an increased expression of IRS-1, this does not result in an increase in Akt phosphorylation or activation of downstream targets of the Insulin/IGF pathway. For this reason, it does NOT appear that GSK-3α has that much of an effect on the Insulin/IGF pathway.
GSK-3α activity is increased by phosphorylation of tyrosine-279 and GSK-3α activity is decreased by phosphorylation of serene-21. Thus upstream “phosphorylators” of GSK-3α can regulate how active this protein is. Unfortunately, I could not find any inhibitors or activators of GSK-3α Most of the work in GSKs has been focused on GSK-3β, which is the “bad GSK”.
Reference:May 2014 GSK-3 as potential target for therapeutic intervention in cancer
b. The “beta isoform” of GSK
– a pro-aging regulator, especially in the brain, the necessary but sometimes bad guy
The strongest data on the fact that GSK-3β is “upstream” in the cause of neurodegenration is the fact that GSK-3β induces both the formation of Amyloid-beta aggregates and also results in the hyperphosphorylation of tau. The molecular mechanism for both of these GSK-3β effects are now well established. No other explanation for AD has been shown to explain both the Amyloid beta part of this disease and the tau hyperphosphorylation part of this disease. GSK-3β expression increases with aging. it appears to be a “pro-aging” pathway that is “upstream” and occurs prior to the formation of the following events and pathologies in the brain”
A. Synaptic dysfunction – Synaptic dysfunction is an “early feature” of AD. It precedes and may even cause the neurodegeneration. Both GSK isoforms are expressed in synapses, especially the hippocampal synaptosomes
B. GSK-3β and NF-kB – GSK-3β phosphorylates NF-kB and promotes the proteasomal degradation of NF-Kb NF-kB is the master of inflammation. It is no surprise then to find out that there are many interactions between GSK-3β and the NF-kB pathway. First of all, GSK-3β directly phosphorylates NF-kB at serene 468. This results in the proteasomal degradation of NF-kB. In addition, GSK-3β interacts with I-kappaB alpha in epithelial cells. This results in the prevention of the degradation of I-kappaB alpha. Thus GSK-3β has an anti-inflammatory effect on the NF-kB pathway. This appears to be an exception to the idea that this GSK-3β isoform is “bad” for aging people.
C. GSK-3β and Wnt signaling – GSK-3β is a central figure in Wnt signaling – it can either increase the degradation of beta-catenin (in unstimulated cells) or increase Wnt/beta-catenin signaling in stimulated cells. The importance of this is treated below when we discuss how GSK-3β is involved in malignant transformations in certain cancers. In the Wnt pathway, scaffolding regulatory proteins control the specificity of GSK-3β mediated signaling. In the absence of cell stimulation, CK1 phosphorylates beta-catenin at S45. This “primes” beta-catenin for subsequent phosphorylation by GSK-3 at S41, S37, and S33. These phosphorylation targets target beta-catenin for ubiquination and proteasomal degradation. The following diagram illustrates this.
Reference: May 2014 GSK-3 as potential target for therapeutic intervention in cancer
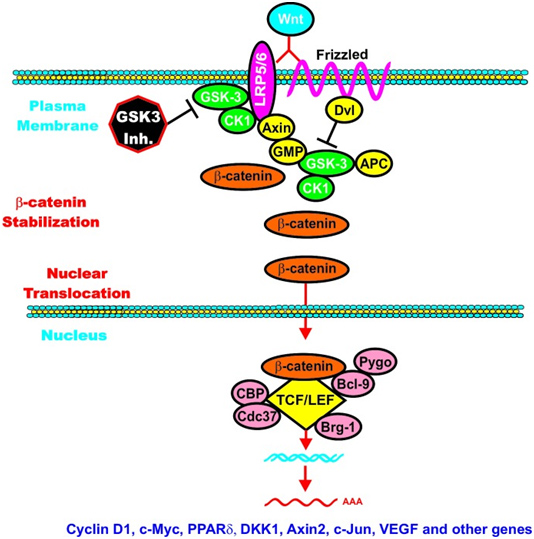
Wnt/beta-catenin induced gene expression is modulated by GSK-3.
However, once the cell is stimulated by Wnt, then the GSK-3 pathway can activate the Wnt-catenin signaling pathway, increasing its activity like an “amplifier”. The molecular mechanism here is the phosphorylation of LRP5/6, which stabilizes Axin. Here is an illustration of this:
Again from Reference: May 2014 GSK-3 as potential target for therapeutic intervention in cancer
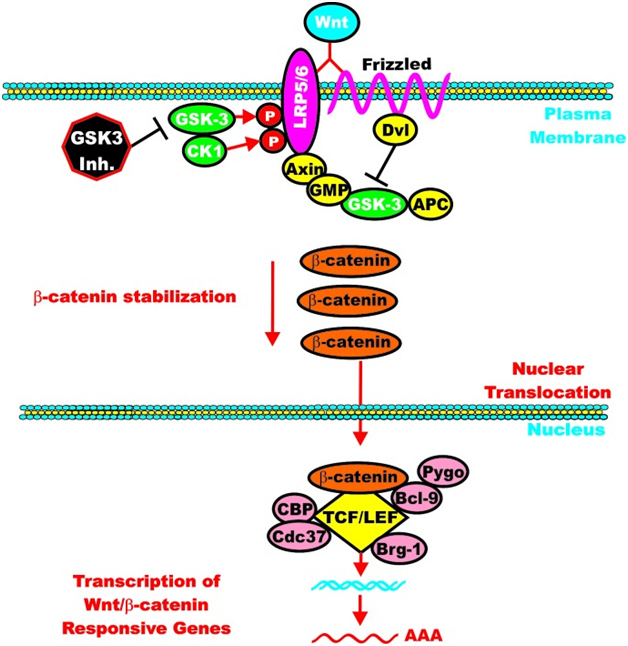
GSK-3 can enhance Wnt/beta-catenin signaling
D. GSK-3β and c-Myc
GSK-3 can regulate c-Myc activity. c-Myc controls cell division and the cell cycle via the CDC25 gene. c-Myc also influences the gene expression of GADD45, the DNA damage pathway that functions as a tumor suppressor by arresting the cell cycle.
Reference:Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7
E. GSK-3β and p53 – There is direct interaction between p53 and GSK-3β after DNA damage to induce apoptosis
After DNA is damaged, the tumor suppressor p53 is the key intermediate that induces either cell cycle arrest or apoptosis of the cell. Apoptosis by p53 after DNA damage is triggered by cysteine/aspartate proteases, such as Caspace-3. However, until recently, there was no explanation for how p53 activated Caspace-3. Then in 2002, Jope and colleagues from UAB showed that with chemotherapy induced DNA damage, the nuclear localized GSK-3β was activated. However, this GSK-3β activation was not due to the normal method of activation, which is the phosphorylation of Tyr-216 on GSK-3β. Instead, there was a direct protein-to-protein interaction between p53 and GSK-3β and this association actuated GSK-3β, making it an active protein that then would phosphorylate other proteins (such as tau).
Reference: 2003Direct, activating interaction between glycogen synthase kinase-3β and p53 after DNA damage
5. Differential GSK Inactivation and Differential Timing of GSK-3 induced activation/inactivation of Transcription Factors
The diagrams above did not include the STAT family of transcription factors. These STAT factors are important because of their roles in oncogenic transformation [“Stats (for signal transducers and activators of transcription) are a family of transcription factors that regulate cell growth and differentiation. Their activity is latent until phosphorylation by receptor-associated kinases. A sizable body of data from cell lines, mouse models, and human tissues now implicates these transcription factors in the oncogenesis of breast cancer(ref). Because Stat activity is modulated by several posttranslational modifications and protein-protein interactions(ref), these transcription factors are capable of integrating inputs from multiple signaling networks. Given this, the future utilization of Stats as prognostic markers and therapeutic targets in human breast cancer appears likely(ref).”] STAT3 and STAT5 are preferentially activated by GSK-3s, whereas STAT1 and STAT6 are not activated very much by the GSK-3s. Although STAT1 is not activated early by certain signaling stimuli (Ex: IFGAR1/2), the same transcription factor can be activated late by GSK-3s. The diagram below illustrates how GSK-3s regulate the STAT family of transcription factors.
Reference for illustration: 2012 Glycogen Synthase Kinase 3: A Point of Convergence for the Host Inflammatory Response
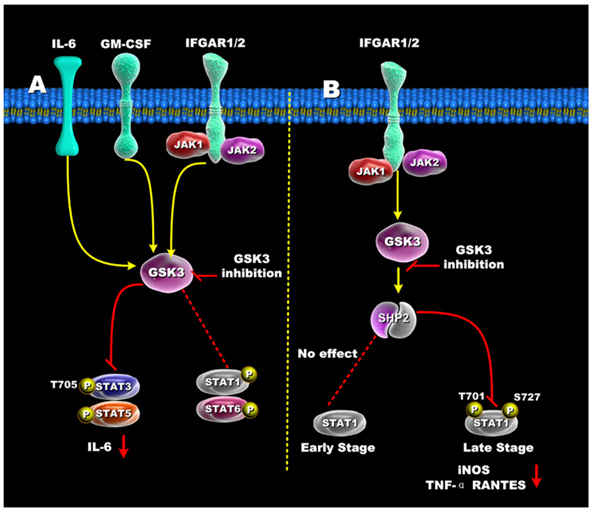
“Regulation of STATs by GSK3 — (A) In IFN-γ, GM-CSF, and IL-6 stimulated cells, GSK3 inhibition suppressed both STAT3 and STAT5 activity, but only minimally affected STAT1 and STAT6 activity. (B) GSK3-β inhibition differentially affects early and late STAT1 activation in IFN-γ-treated cells. Inhibition of GSK3 had no effect on early STAT1 activity (<1hr) but diminished late STAT1 phosphorylation by increasing SHP-2 activity. The ability of GSK3 inhibition to reduce STAT1 activity resulted in reduced levels of iNOS, TNF, and RANTES by IFN-γ-stimulated macrophages.”
6. The Four Ways that GSK-3 activity is regulated
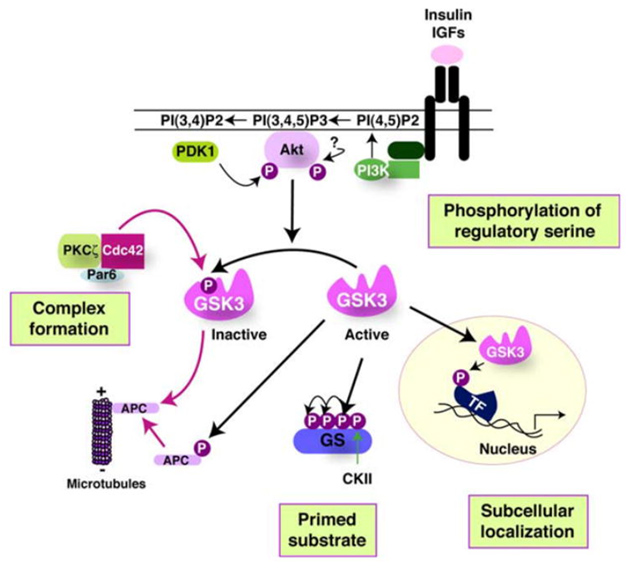
The activity of GSK-3s is highly regulated. Four key mechanisms have been identified that regulate GSKs. They are
1)The phosphorylation of GSK3 itself, 2) the sub cellular localization of GSK3s, 3)The formation of protein complexes containing GSK3s, and 4)the phosphorylation state of the GSK3 substrates. The diagram below illustrates these 4 regulatory mechanisms:
Reference 2007 Glycogen Synthase Kinase-3 (GSK-3): Inflammation, Diseases, and Therapeutics
7. Primed vs Non-primed sustrates
Active GSK-3s exhibit a 100 to 1,000-fold increase in substate specificity for pre-primed substrates, compared to non-primed substrates. N-terminal phosphorylation of GSK-3β (Ser9) acts as a “pseudosubstrate” for the phosphate binding site and thus competes for the binding of arginine 96 with pre-primed, vs non-primed substrates. The diagram below shows what this effect does with pre-primed substrates.
Reference: Glycogen Synthase Kinase 3: A Point of Convergence for the Host Inflammatory Response
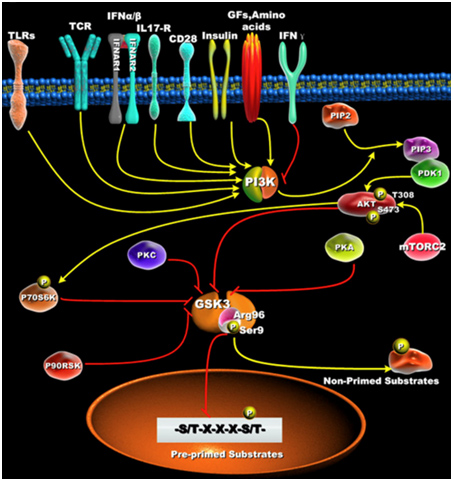
“ — it is first important to define GSK3’s preference to phosphorylate substrates that are pre-primed (pre-phosphorylated) on a serine or threonine located around a five amino acid consensus sequence corresponding to serine/threonine-X-X-X-serine/threonine-P, where the first serine or threonine is the residue to be phosphorylated by GSK3, X can be any amino acid, and the last serine- or threonine-P is the pre-primed (pre-phosphorylated) residue [42](Figure 1B). The preferential phosphorylation of pre-primed (pre-phosphorylated) substrates by GSK-3 has been shown to result in increasing substrate phosphorylation efficiency by more than 100 to 1000-fold, as compared to non-primed substrates [43].”
8. GSK-3S and Aging – Is it Due to Alpha or Beta?
There is a lot of confusion as to whether GSK-3s are “pro-aging” or “anti-aging” factors. The answer is “both”. It is becoming clear that GSK-3s are both the activators of aging (GSK-3 beta) and the inhibitors of aging (GSK-3α). GSK-3α and GSK-3β are 98% identical in their kinase domains, but due to their N-terminal and C-terminal ends, their effects on aging are the opposite. The effect of GSK-3s on downstream target proteins is their ability to phosphorylate the “downstream target” protein. Phosphorylation of the GSK 3 target usually inactivates it and may in some cases, result in the proteasomal degradation of the down stream target protein (i.e. by the UPS system). Unlike most protein kinases, GSKs are typically active in unstimulated cells and when the cell is activated, the GSKs are inhibited.
9. Inhibition of GSK-3 β signaling by lithium
Most importantly, there is something that we can do about GSK 3β signaling (the pro-aging form). Lithium has been known to increase activator protein 1 (AP-1) binding to DNA, but the exact mechanism of action eluded researchers for a long time. Once GSK-3β was discovered, the mystery was solved. GSK-3β phosphorylates c-Jun (JNK). This JNK phosphorylation prevents activator protein 1 (AP-1) from binding to DNA. As a result, since Lithium inhibits GSK-3β, this inhibition resulted in an increase in AP-1 binding to DNA, thus activating many genes. Lithium increases activator protein 1 (AP-1) binding to DNA by inhibiting GSK-3β, which then cannot phosphorylate JNK, which then cannot inhibit AP-1 binding to DNA. In Item 10 below, we describe how lithium inhibition of GSK-3β also extends concentrations of Nrf2 by preventing phosphorylation of Nrf2 by GSK-3β.
References:
- 2009Validating GSK3 as an in vivo target of lithium action
- 2003 Lithium blocks the c-Jun stress response and protects neurons via its action on glycogen synthase kinase 3
Lithium carbonate or Lithium chloride, compounds that have been used since 1871 by psychiatrists for treating mania, bipolar illness, and depression. The FDA approved Lithium in 1970 and although most patients have now been treated with other medications for depression and bipolar illness, it is still available and is inexpensive. Lithium has already been proven to prolong lifespan in all lower animals. Lithium is a safe, clinically effective inhibitor of GSK 3β. At high levels used for treating psychiatric conditions, there are a lot of side effects. However, at lower concentrations needed to inhibit GSK-3s, there are very few side effects. The following facts support this emerging view that Lithium is a longevity agent that works via GSK-3 inhibition and promotes longevity and decreased neurodegeneration.
A. Low dose lithium uptake promotes longevity in humans and metazoans – A large Japanese cohort study was done and reported in 2011 which showed that tap water-derived lithium and a longevity effect. There was an inverse correlation between drinking water lithium concentrations and all cause mortality in 18 different Japanese municipalities with a total of 1,206,174 people (p = 0.003). These same authors also showed that low dose lithium exposure of lithium chloride extended life span in C elegans (p = 0.047).
Reference: 2011 Low-dose lithium uptake promotes longevity in humans and metazoans
B. Lithium administration increases brain gray matter – Several studies of patients with bipolar illness who were treated with Lithium showed dramatic, measurable, statistically significant increases in brain gray matter. (p = 0.0043)
References:
- 2009 A longitudinal study of the effects of lithium treatment on prefrontal and subgenual prefrontal gray matter volume in treatment-responsive bipolar disorder patients
- 2010 Lithium-induced gray matter volume increase as a neural correlate of treatment response in bipolar disorder: a longitudinal brain imaging study
- 2002 Increased gray matter volume in lithium-treated bipolar disorder patients
- 2010 Lithium-induced gray matter volume increase as a neural correlate of treatment response in bipolar disorder: a
C. Lithium in tap water reduces suicide mortality – A large study of 40 different municipalities in Japan showed that there was an inverse relationship between suicide and tap water lithium levels in the Aomori prefecture. The Aomori prefecture has the highest rates of suicide mortality in Japan. This study suggests that the low dose lithium in tap water may protect those who drink tap water from depression, which is the leading reason why Japanese commit suicide.
Reference: 2011 Low-dose lithium uptake promotes longevity in humans and metazoans
10. Inhibibiting GSK-3β enhances expression of Nrf2 since GSK-3β phosphorylates Nrf2.
Many of the health effects of GSK-3β inhibition and the use of lithium may be due to this effect. See the 2012 publication Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/β-TrCP axis “Here, two-dimensional (2D) gel electrophoresis and site-directed mutagenesis allowed us to identify two serines of Nrf2 that are phosphorylated by glycogen synthase kinase 3β (GSK-3β) in the sequence DSGISL.” And see the 2013 publication Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity, Supression of GSK-3β signaling could also explain why lithium works in part by activating Nrf2 signaling See the just published report Antiarrhythmic effect of lithium in rats after myocardial infarction by activation of Nrf2/HO-1 signaling. “In conclusions, lithium protects ventricular arrhythmias by attenuating NGF-induced sympathetic innervation via antioxidant activation of Nrf2/HO-1 axis.” We have written extensively in this blog about the important health benefits of the Nrf2 pathway(ref)(ref)(ref).
An important corollary of this could be that phytochemical activation of Nrf2 could be frequently or even generally mediated by inhibition of GSK-3β. At least this appears to be the case for the phytochemical nordihydroguaiaretic acid. See the 2012 publication Signaling pathways activated by the phytochemical nordihydroguaiaretic acid contribute to a Keap1-independent regulation of Nrf2 stability: Role of glycogen synthase kinase-3. “Our study demonstrates that NDGA activates Nrf2 through multiple signaling cascades and identifies GSK-3β as an integrator of these signaling pathways and a gatekeeper of Nrf2 stability at the level of the Neh6 phosphodegron.” We want to do further research and check out whether this corollary is also true for the many other phyto substances which activate Nrf2. In Vince’s treatise he identified 36 of these. If the corollary is generally correct, it might explain the puzzling issue of how phytochemicals upgrade the expression of Nrf2.
11. GSK 3s and Inflammation – Diabetes, Cancer, Multiple Sclerosis, Alzheimers, and Colitis
It is now clear that Glycogen Synthase Kinase 3 plays an important role in several diseases via their ability to up regulate inflammation. GSK-3s are powerful regulators of inflammation because of their ability to act as a “kinase cascade amplifier”. As a result, GSK-3s have been found to play an important role in the CNS neuroinflammatory diseases such as multiple sclerosis, Alzheimer’s disease, and mood disorders. Here the cells that are involved include microglia and astrocytes. GSK-3s also play a key role in peripheral inflammatory disease such as colitis and arthritis via macrophages/monocytes. GSK-3s also appear to play a role in Diabetes via their local inflammation effect and in Cancer via their general inflammation effect. The diagram below illustrates this:
Reference for diagram:2007 Glycogen Synthase Kinase-3 (GSK3): Inflammation, Diseases, and Therapeutics
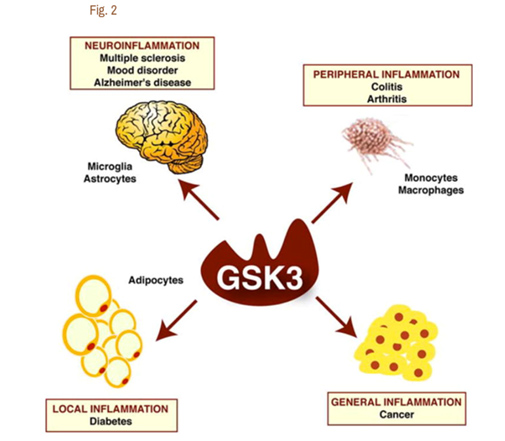
A. GSK-3s and TLR-induced inflammation – Since GSK-3s have such a vital role in inflammation, GSK-3 inhibitors have been shown to have a very strong anti-inflammatory effect on a cellular level. The brain normally is protected from the classical immune inflammatory response, however. This is probably one of the most important roles of the blood brain barrier (BBB) and is why the brain has been referred to as “immune privileged”. Unfortunately, when the BBB is disrupted or inflamed, macrophages or monocytes invade the brain and activate resident glia cells, including astrocytes and microglia. This is why with many neurodegenerative disorders, gliosis is a sine qua non of the disease on histology. The primary way that GSK-3s amplify inflammation is in the toll-like receptor (TLR) induced inflammation. Here are the classic TLR receptor and what activates them:
TLR receptor subtype What activates them
- TLR2 lipoteichoic acid from Streptococcus pneumonia
- TLR4 synthetic lipid A
- TLR5 flagellin protein from Salm typhimurium
- TLR9 human CpG
When any of the above TLR agonists are used to stimulate human peripheral mononuclear cells (PBMCs) in the presence of a GSK-3 inhibitor, there was a selective reduction of 50-90% in the pro-inflammatory cytokines IL-1β, IFN-γ, IL-12, and IL-6. The following paragraph goes into more detail on this.
Reference:2012 Glycogen Synthase Kinase 3: A Point of Convergence for the Host Inflammatory Response
B. GSK-3s and cytokine-induced inflammation – Recent work has shown that GSK-3 activity is required for the full stimulation of the production of several pro-inflammatory cytokines. This includes IL-6, IL-1beta, and TNFalpha, following stimulation by toll-like receptors in monocytes and peripheral blood mononuclear cells. GSK-3s also modulate the production of the anti-inflammatory cytokine, IL-10. When a GSK-3 inhibitor was used in vitro, the production of IL-10 increased by 3-5 fold in human monocytes activated with LPS. On the contrary, GSK-3s increase the production of the pro-inflamatory cytokine, IL12. When a GSK-3 inhibitor was used in vitro, it reduced the production of IL-12 by LPS-stimulated human monocytes by 70%. With GSK-3 inhibitors, IL-6 and TNF-α production by LPS-stimulated monocytes was reduced by 60-80%.
C. GSK-3 isoforms are differentially activated with inflammation – Despite these harmful effects of GSK-3s, they are vital to life. Deletion of the GSK-3β gene results in death of the embryo. Adding GSK-3α is unable to “rescue” the GSK-3β “knock out” mice. When probes were used to determine which GSK-3 was involved with inflammation, it was clear that GSK-3β was activated via the phosphorylation of Ser-9 by LPS-induced inflammation, whereas GSK-3α was not activated with LPS-induced inflammation.
References:
2007 Glycogen Synthase Kinase-3 (GSK3): Inflammation, Diseases, and Therapeutics
12. GSKs and Alzheimer’s disease
As we mentioned above, the GS-3s induce the phenomena thought to be the cause of Alzheimer’s disease according to both of the leading theories: the beta amyloid theory(ref) and the tau tangles theory(ref), since they promotes the expression of both beta-amyloid and tau tangles. In fact, GSK-3B is that it is the only protein that directly cause all of the following features of AD
- GSK-3 is increased in white cells early in Alzheimer’s disease.
Reference: Glycogen synthase kinase-3 is increased in white cells early in Alzheimer’s disease
- GSK-3beta induces memory deficits in vivo, long before the appearance of Amyloid-beta aggregates and before the appearance of tau tangles. Reference: GSK-3 is essential in the pathogenesis of Alzheimer’s disease
- GSK-3alpha induces Amyloid beta productionand GSK-3alpha inhibition reduces GSK-3alpha reduces Amyloid beta production. Reference: GSK-3 is essential in the pathogenesis of Alzheimer’s disease
- Amyloid beta fragments (Abeta 25-35) activate tau protein kinase I(TPKI). Reference: Activation of tau protein kinase I/glycogen synthase kinase-3beta by amyloid beta peptide (25-35) enhances phosphorylation of tau in hippocampal neurons
- Tau protein kinase 1 (TPKI) is the same thing as Glycogen Synthase Kinase 3beta (GSK-3B). Thus, GSK-3b IS THE ENZYME THAT ACTUALLY DOES THE HYPERPHOSPHORYLATION! References: Physiology and pathology of tau protein kinases in relation to Alzheimer’s disease, Alzheimer’s disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells
Synaptic dysfunction – Synaptic dysfunction is an “early feature” of AD. It precedes and may even cause the neurodegeneration. Both GSK isoforms are expressed in synapses, especially the hippocampal synaptosomes.
A very interesting angle is the role of GSK-3 in the relationship of H. pylori to Alzheimer’s disease. A July 2014 publication Helicobact er pylori Filtrate Induces Alzheimer-Like Tau Hyperphosphorylation by Activating Glycogen Synthase Kinase-3β draws a connection: “Abnormal hyperphosphorylation of microtubule-associated protein tau is involved in the pathogenesis of several neurodegenerative disorders including Alzheimer’s disease (AD). Helicobacter pylori (H. pylori) infection has been reported to be related with a high risk of AD, but the direct laboratory evidence is lacking. Here we explored the effect of H. pylori infection on tau phosphorylation. The results showed that H. pylori filtrate induced significant tau hyperphosphorylation at several AD-related tau phosphorylation sites, such as Thr205, Thr231, and Ser404, both in mouse neuroblastoma N2a cells and rat brains with activation of glycogen synthase kinase-3β (GSK-3β). Application of GSK-3 inhibitors efficiently attenuated the H. pylori-induced tau hyperphosphorylation. Our data provide evidence supporting the role of H. pylori infection in AD-like tau pathology, suggesting that H. pylori eradication may be beneficial in the prevention of tauopathy.”
13. GSK-3s and cancer
Aberrant activity of GSK-3 has been implicated in many types of cancers, especially those that are resistant to chemotherapy, radiation therapy, and targeted therapy. Targeting GSK-3 may be a means to overcome the resistance of these cancers to certain chemotherapeutic drugs, radiation, and small molecule inhibitors. Here are some example cancers where GSK-3 is known to be involved with carcinogenesis:
A. GSK-3 and beta-catenin in liver cancer – decreased GSK-3 levels due to HBx and ERK, HCV core protein GSK-3β is known to be involved with the formation of liver cancer. The dysregulation of GSK-3β phosphorylation and inhibition of GSK-3β activity and beta-catenin signaling is the molecular mechanism involved with this type of cancer. In a recent study of 80 patients with hepatocellular cancer, the expression of GSK-3β protein was noted to be decreased. However, exceptions to this trend were also found. In some cancers, GSK-3β was noted to have high basal levels of S9-phosphorylated GSK-3β.
B. HBx and ERK – The molecular mechanism by which GSK-3β levels are decreased in hepatocellular carcinoma appears to be related to one of the hepatitis virus proteins which is expressed in liver cells in the precancerous phase of liver cancer. This virally produced protein is called HBx. HBx activates ERK. This “primes” ERK for GSK-3β phosphorylation. ERK phosphorylates GSK-3β at T43. Then p90Rsk phosphorylates GSK-3β at S9, which then results in the inactivation of GSK-3β. This inactivation of GSK-3β results in beta-catenin stabilization and signaling. Similar molecular mechanisms occur which stabilizes paxillin, a cytoskeleton regulator.
C, HCV protein – Another way that GSK-3s are involved with hepatocellular carcinomas involves the HCV core protein. HCV core protein increases in hepatic cancers and stabilizes beta-catenin levels. This occurs via the inactivation of GSK-3β by the phosphorylation of GSK-3β at Ser-9. Once beta-catenin is stabilized, then it can enhance canonical Wnt/beta-catenin targets, such as c-Myc, cyclin D1, Wnt 1-inducible signaling pathway protein 2 (WISP2), and connective tissue growth factor (CTGF). All of these genes are up regulated when beta-catenin stabilized by the GSK-3 mediated HCV core protein.
D. SIRT2 upregulation – GSK-3β and beta-catenin signaling plays a role in the Sirtuin 2 mediated transformation of liver cells into hepatocellular cancer. SIRT2 is up regulated in about 50% of hepatocellular cancers. In these cancers, there is a shorter over all survival. SIRT2 regulates the activation of Akt by deacetylating Akt. Once Akt is deacetylated, it can then phosphorylate GSK-3β and inactivate the protein. This activates the beta-catenin pathway and promotes cell migration of hepato-cellular carcinoma cells. For this reason, many have suggested that GSK-3β is a “tumor suppressor” and that the loss of GSK-3β expression or activity may contribute to hepatocellular cancer development. Thus, there appears to be a very good side to GSK-3β in addition to all the evil sides we have been describing.
E. Cancer treatment with GSK-3β inhibitors – increased survival in a Wnt/beta-catenin independent mechanism. Studies of GSK-3 inhibitor treatment of cancer cells have shown that cancers are inhibited with these compounds. This is a paradoxical finding, since an “inhibitor of a tumor suppressor” should make the cancer cells grow. Instead, the cancer cells show a reduced survival and proliferation via a Wnt/beta-catenin independent mechanism. Specifically, treatment of cancer cells with a GSK-3β inhibitor decreases telomerase expression (hTERT) and also reduces telomerase activity. This occurs with both non-selective inhibitors such as Lithium and with selective inhibitors such as SB-415286. Here their anti-cancer effects are mediated by apoptotic signaling (Caspace-8, caspace-3, and p53 gene activation).
Conclusion: Hepatocellular cancer development can be explained in part by the down-regulation of GSK-3β activity by HBx/ERK mechanisms, by HCV protein mechanisms, and by SIRT2 dependent mechanisms. These studies suggest that GSK-3β functions as a tumor suppressor and that an inhibitor would have an adverse effect on cancer, causing the cancer to proliferate. However, the opposite effect occurs when GSK-3 inhibitors are used on cancer cells. Instead, the cancers show reduced survival and proliferation via Wnt/β-catenin independent mechanisms involving hTERT and apoptotic proteins. Again, an example of how interacting pathways sometimes work to produce unexpected results.
Thus, GSK-3 inhibitors show promise in the treatment of cancer.
Reference: May 2014 GSK-3 as potential target for therapeutic intervention in cancer.
14. GSK-3s and signaling in selected pathways
The GSK-3s impact on a number of pathways that are involved both in embryogenesis and cancer processes, We comment on some of these.
A. GSK-3 and mTORC1 signaling – AMPK can down-regulate mTORC1 via GSK-3 induced TSC2 degradation. However, Wnt signaling can up-regulate the mTORC1 pathway via GSK-3 via beta-catenin mediated inhibition of TSC2 degradation. GSK-3 always prefers “primed substrates.” This is true for the tuberous sclerosis complex 2 (TSC2) in the mTOR pathway. Specifically, AMPK can act as the “primer” and first phosphorylates TSC2. Then GSK-3 will phosphorylate TSC2 and thus activate the proteasomal degradation of TSC2. This results in the down-regulation of the mTOR pathway. In contrast, Wnt signaling suppresses the GSK-3 mediated phosphorylation of TSC2. This results in the activation of the mTOR pathway. Rapamycin inhibits mTORC1 and this then suppresses the Wnt/beta-catenin induced cellular proliferation of cancer. This is how GSK-3 plays a role in cancer via the mTORC1 pathway. In addition to GSK-3, three other proteins are involved in the modulation of the effects of Wnt signaling on cancer (DKK1, Dvl, and Axin).
B. GSK-3 and SMAD1 – MAPKs can “prime” SMAD1 and then GSK-3 can activate the “primed” SMAD1 transcription factor. The SMAD1 transcription factor is the downstream transcription factor that mediates the effects of TGF-beta superfamily ligands such as BMPs. SMAD1 signaling is often increased in cancer. As mentioned before, GSK-3 prefers “primed substrates”. In this case MAPKs such as ERK, JNK, and p38 can each phosphorylate SMAD1. Then GSK-3 will phosphorylate SMAD1 and thus induce its ubiquitinylation and proteasomal degradation. As seen with the other examples above (TSC2), Wnt signaling suppresses the GSK-3 mediated phosphorylation of SMAD1 and therefore prevents the proteasomal degradation of SMAD1. This system of regulation plays an important role in embryonic pattern formation during embryogenesis.
C. GSK-3 and Hedgehog Pathway – GSK-3 plays a role in segmental pattern formation in the embryo. The Hedgehog pathway is very important in development (embryogenesis), but is normally silenced in most adult tissues. However in cancer, the Hedgehog pathway can be re-activated. The classic cancers where the Hedgehog pathway is re-activated are basal cell carcinoma of the skin and medulloblastoma. Here constitutive Hedgehog pathway activation occurs due to mutations in various genes of the Hedgehog pathway such as the receptor, patched 1 (PTCH1) or smoothened (SMO). Other types of Hedgehog signaling occurs in cancer too. One of the most common is paracrine Hedgehog signaling. This occurs in a subset of epithelial cancers found in the colon, panaceas, and ovaries. Here the cancer cells secrete Hedgehog ligands which then activate Hedgehog signaling in the surrounding non-cancerous stroma cells, creating a “cancer favorable environment” for the cancer cells to grow and then for the cancer cells to invade the stroma (i.e. local recurrence of cancer).
Most of what we understand about the Hedgehog pathway comes from studying embryology. During embryogenesis, the Hedgehog pathway regulates segmental pattern formation via three genes: desert hedgehog (DHH), Indian hedgehog (IHH), and sonic hedgehog (SHH). Hedgehog ligands bind to their receptor, PTCH1. This causes the internalization and degradation of smoothened (SMO), which then promotes the dissociation of SUFU-glioma associated oncogene homolog (GLI). GLI3 can be phosphorylated by PKA, GSK-3 and CK1. Once GLI3 is phosphorylated, it becomes a transriptional repressor. Activatee GLI1 proteins stimulate the transcription of Hedgehog target genes, which includes IGF2. IGF2 has been shown in some cancers to induce PI3K/PTEN/Akt/mTOR pathway and thereby increase cancer cell proliferation and survival. In addition, IGF2 can also inhibit GSK-3β. GSK-3 can positively regulate hedgehog signaling. When GSK-3 is activated, the hedgehog signaling pathway is activated. When GSK-3 is inactivated, then the hedgehog signaling pathway is promoted. GSK-3 affects hedgehog signaling pathway via the phosphorylation of SUFU, a binding partner for GSK-3β.
D. GSK-3 and Notch Signaling Pathway – GSK-3 regulates notch signaling, EBV viral protein EBNA2 is a “biological equivalent” of a Notch receptor. Notch signaling plays an important role in a wide variety of cancers. The prototypical cancer where Notch signaling is unregulated is T cell acute lymphoblastic leukemia (T-ALL). In T-ALLs, 50% of cancers have Notch signaling up regulated. However, it is also up regulated in some breast cancers, cervical cancers, pancreatic cancers, endometrial cancers, renal cancers, lung cancer, colon cancer, head and neck cancer, Hodgkin’s lymphoma, and large cell anapestic lymphoma. The most common way that Notch Signaling is up regulated in cancer is the over-expression of the receptor gene.
The classic form of Notch 1 receptor over expression is seen in cervical cancer. 99% of cervical cancers are associated with high risk HPV virus. The viral oncogenes E6 and E7 disrupt the cell cycle regulation by targeting p53 and Rb. However, the E6 and E7 disruption of the cell cycle control by p53 and Rb is not enough to transform the cervical epithelial cells into cancer cells. To do this, the Notch 1 receptor must be over-expressed. As the pre-malignant cells progress farther and farther towards becoming cancer cells, the expression of Notch 1 receptor gradually increases. It is not surprising when researchers found out that the up regulation of the Notch 1 receptor was induced by E6 and E7 viral oncogenes.
There is another small class of tumors that appear to be caused by the Epstein Barr virus where Notch signaling has a very pivotal role. These cancers include Burkitt’s lymphoma, Hodgkin’s lymphoma, nasopharyngeal carcinoma, and lymphomas in immune compromised patients. The EBV can “immortalize” B lymphocytes and the process of B cell immortalization is a EBV protein called the “transactivator EBNA2”. EBNA2 controls the expression of several viral genes and cellular genes. EBNA2 is tethered to promoter regions of these genes by a cellular repressor called CSL. CSL resembles the physiological activation of CSL-repressed promoters by intracellular Notch receptors. Thus, the EBNA2 protein is the “biological equivalent” of an intracellular Notch receptor (N-ICD). EBNA2 and N-ICD have been shown to be partially interchangeable.
The primary way that the Notch Signaling Pathway is activated is by direct cell-to-cell contact. One cell has what is called a “ligand” and the other cell has a “receptor” that matches the ligand. The ligand-receptor interaction is what triggers the Notch signaling pathway. There are six ligands that have been identified in human cells (DII1, DII3, DII4, Dik, Jagged1, and Jagged 2). There are four transmembrane receptors found in the Notch signaling pathway (Notch 1, Notch 2, Notch 3, and Notch 4. The cell-to-cell contact and binding of a Notch ligand to a Notch receptor (DII2, etc.) then can trigger a number of intracellular pathways, such as the Ras/MAPK pathway, the PKC/NF-kB pathway, the Wnt signaling pathway, the TGF-beta pathway, and the Sonic Hedgehog pathway (Shh). All of these pathways promote tumor growth. In the Shh pathway, GSK-3β phosphorylates NICD, This phosphorylation event results in the prevention of the proteasomal degradation of NICD. This results in the prolonged survival of the NICD protein by preventing its proteasomal degradation.
Reference: May 2014 GSK-3 as potential target for therapeutic intervention in cancer
E. GSK-3β and Tumor cell Exosome Signaling –Tumor cells secrete exosomes rich in lipid-raft containing membranes that interact with other tumor cells by using Notch signaling to increase PTEN and GSK-3β expression in the target cell that the exosomes fuse with. Another very interesting study of pancreatic cancer cells has recently been done, focusing on the role of secreted exosomes from the cancer cells. Exosomes are “nano particles” made by the endosomal compartment of the cell. These secreted nano particles contain both cell surface ligands that allow for docking onto the surface of other cells and surface ligands that trigger endocytosis by the cell that does the “receiving”. This exosome/cell interaction led to the decreased expression of a nuclear Notch receptor target called Hes-1. Unexpectedly, blocking presenilin resulted in the activation of PTEN and GSK-3β. Conversely, inhibiting either PTEN or GSK-3β resulted in the increase in He’s-1 expression. This inhibition increased the apoptotic response and decreased the survival of the cancer cells.
Reference: 2009, Essential role of Notch signaling in apoptosis of human pancreatic tumor cells mediated by exosomal nano particles
Cancer and GSK-3β – Conclusions: GSK-1s play a major role in cancer by their interaction with the Wnt/beta-catenin pathway, the mTOR pathway, SMAD1, the Hedgehog pathway, and the Notch pathway. In most all of these pathways, another kinase first primes the protein by phosphorylating the GSK-1 target first, then GSK-1beta phosphorylates the target protein. This results in the proteasomal degradation of the protein that has been phosphorylated. However with cancer, there is a dysregulation in GSK-3β.
15. GSK-3β inhibitors – compounds and drugs that may help prevent or treat Alzheimer’s disease
There are a large number of compounds that have been shown to inhibit GSK-3s. The way they have been found is by In vitro testing of compounds to see if they can activate GSK-3 inhibitor, the effects of these other GSK-3 inhibitors is additive. This means that the two together have a greater effect than either one could achieve alone (i.e. insulin + a GSK inhibitor). The GSK-3 inhibitors that have been found so far include metal cations, aminopyrimidines, arylindolemaleimide, thiadiazolidindiones, halomethylketones, peptides, and natural products derived from marine organisms. Here is a partial list of these compounds:
A. Metal Cations – non-selective, ATP noncompetitive inhibitors
We have already discussed lithium, the most important of these. Several other divalent metal cations also inhibit GSK-3 by their divalent positive charge. These include Beryllium, Copper, Mercury, and Tungsten. Lithium appears to be the most selective GSK-3β inhibitor of this group of metal cations, however. Unfortunately, these metal cations are NOT selective inhibitors of GSK-3. For instance, Lithium chloride or Lithium carbonate will also inhibit casein kinase-2, p38 regulated/activated kinase, and MAPK activated protein kinase-2. Lithium is also an inhibitor of polyphosphate 1-phosphatase and inositol monophosphate, two enzymes that are required for the synthesis of inositol. Paradoxically, Lithium chloride has also been reported to acutely elevate phosphatidylinositol 3-phosphate levels in rat cerebellar granule cells, thereby activating PKB. It is this “non-selectivity” problem that has prevented the widespread use of Lithium chloride for other diseases other than depression and bipolar illness.
Many of the other metal cations mentioned above also have serious toxicity, such as Copper and Mercury. For this reason, these other metal cations are not likely to become “drugs” anytime soon.
References:
- 2005 GSK-3 activity in neocortical cells is inhibited by lithium but not carbamazepine or valproic acid
B. Valproic acid
Sodium valproate is also a weak inhibitor of GSK-3β. Valproic acid increases activator protein 1 (AP-1) binding to DNA, but only at much higher concentrations than lithium. The direct mechanism of how valproic acid works is similar to how lithium works. At high concentrations, valproate can inhibit GSK-3β, and therefore activate SP-1 induced gene expression. However, there is some debate as to whether clinical levels of valproate directly inhibits GSK-3β. Clinically, It may be that Valproic acid inhibits GSK-3β by some indirect mechanism.
References:
- For general information on valproic acid see the 2010 blog entry Valproic acid – The phoenix drug arises again
- 1999 The mood-stabilizing agent valproate inhibits the activity of glycogen synthase kinase-3
- 2002 Valproate regulates GSK-3-mediated axonal remodeling and synapsin I clustering in developing neurons
- 2005 GSK-3 activity in neocortical cells is inhibited by lithium but not carbamazepine or valproic acid.
- 2005 Opposite effects of lithium and valproic acid on trophic factor deprivation-induced glycogen synthase kinase-3 activation, c-Jun expression and neuronal cell death
C. Aminopyrimidines
These are synthetic compounds:
- CT98014
- CT98023
- CT99021
- TWS119
C. Thiadiazolidindiones
One of the FDA approved GSK-3β inhibitors is Rosiglitazone, a drug with a somewhat rocky history.
D. Maleimide derivatives – SB-213763, SB-415286
Maleimide derivatives were identified as leads from a high throughput screen of SmithKline Beecham compound bank against rabbit GSK-3α. SB-213763 is an arylindolemaleimide with an IC50 of 34 nM. SB-415286 is a anilinomaleimide with an IC50 of 78 nM. Both of these are compounds that inhibit GSK-3α in a ATP competitive manner. The two compounds were equally effective at inhibiting human GSK-3α and human GSK-3β. The additional good feature of these molecules is that they are very selective kinase inhibitors and showed little or no inhibition of 24 other kinases when tested against these other kinase compounds in vitro. The maleimide derivatives have an “additive effect” with insulin to stimulate glycogen synthesis. Unfortunately, the maleimide derivatives do not have any synergistic effects with other GSK-3 inhibitors, such as Lithium chloride.
E. ATP-competitive compounds
F. Natural products from Marine organisms
Several compounds have been found from marine organisms that inhibit GSK-3. They include the following:
- 6-BIO
-
Dibromocantharelline
- Hymenialdesine
- Indirubins
- Meridianins
G. Halomethylketones
- HNK-32
- H. peptides
16. Inhibition of GSK-3β may remove the “ceiling” of the cardioprotective effects of certain phytochemicals in postconditioning after ischemia
At least, this appears to be the case for genistein, a soy derivative. The 2009 publication The Ceiling Effect of Pharmacological Postconditioning with the Phytoestrogen Genistein Is Reversed by the GSK3β Inhibitor SB 216763 [3-(2,4-Dichlorophenyl)-4(1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione] through Mitochondrial ATP-Dependent Potassium Channel Openingreports: “In conclusion, the cardioprotective effect of pharmacological postconditioning with genistein and 17β-estradiol involves mitochondrial KATP channels and is limited by a ceiling effect of protection. This loss of cardioprotection is associated with a loss of Akt phosphorylation capacity with increased durations of ischemia. However, this ceiling effect can be reversed by administration of the GSK3β inhibitor SB 216763 through the opening of mitochondrial KATP channels.” This publication raises several interesting issues such as: 1. The roll of the mitochondrial KATP channels in post and possibly pre-conditioning, 2. Whether other phytochemicals might exercise similar effects possibly employing other GSK-3β inhibitors such as lithium, and 3. Whether an enhanced pre-conditioning and post-conditioning effect can thus be realized for other forms of trauma such as surgery, head injuries, etc.
17. On scarcity of beta-catenin, impact of this on GSK3s, and missed opportunities for healh enhancement
The 2013 publication “Gone with the Wnts: Beta-catenin, T-Cell Factor, Forkhead Box O, and Oxidative Stress in Age-dependent Diseases of Bone, Lipid, and Glucose Metabolism” introduces an additional complication regarding health and longevity interventions based on GSK-3.Here is the diagram from that article:
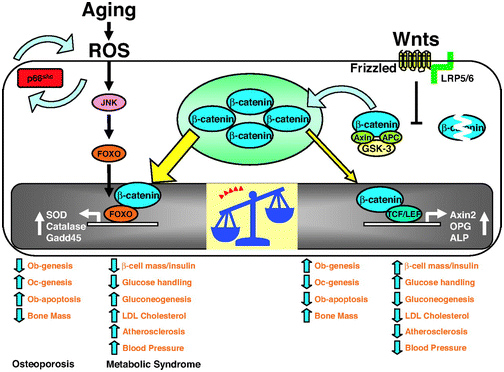
We mentioned above that GSK-3α inhibits Wnt signaling by phophosphorylating beta-catenin, which targets it for ubiquitin proteasomal degradation. Further that WNT signaling can be a “pro-aging pathway,” and we discussed its role in cancer processes.
Normally, Wnt signaling induces positive effects on bone, the pancreas, and lipids. Specifically, Wnt activation of the LRP5/6 Frizzled receptor induces inactivation of GSK-3β. In addition, with plenty of beta-catenin in the nucleus, beta-catenin can act as a cofactor to activate the “downstream target genes of Wnt signaling”, which are mediated by the transcription factor,TCF/LEF. This results in the activation of Axin2, OPG, ALP, and other genes that produce the following phenotype:
Osteoblast stimulation and prevention of apoptosis => bone mass maintenance
- Osteoclast inhibition => no osteoporosis
- Beta-cell mass maintenance in the pancreas => normal insulin signaling
- Decreased gluconeogenesis by the liver => less non-dietary, hepatic-induced hyperglycemia
- Decreased LDL synthesis by the liver => lower LDL cholesterol levels => less oxidized LDL signaling via the Lectin-like LDL receptor of the cells => less p66shc activation
- Less atherosclerosis
- Lowered blood pressure
Unfortunately, there are other transcription factors that requires beta-catenin as a cofactor. These are the Forkhead Box transcription factors (FoxOs). FoxOs need beta-catenin as a cofactor for activation of the genes involving oxidative stress signaling. Thus FoxOs ”steal” beta-catenin away from TCF/LEF. This “shift to the left” of beta-catenin supply results in a change from the “beneficial effects” of Wnt signaling to the “adverse effects” of Wnt signaling.
The nuclear translocation of FoxO transcription factors (FoxO1, FoxO3a, FoxO4, and FoxO6) normally occurs by the inhibition of the Insulin/IGF-1 pathway, since Akt phosphorylation prevents the nuclear localization of FoxOs. However, nuclear translocation of FoxOs can all be triggered by ROS, by one of the MAPK kinases called c-Jun (JNK). In both scenarios, beta-catenin is required as a cofactor for the activation of genes required for cellular resistance to oxidative stress and DNA damage. (SOD, Catalase, Gadd45, etc.)
Conclusion: in the presence of oxidative stress, there can not be enough nuclear beta-catenin to “go around”. As a result, there is inadequate stimulation of the downstream targets of Wnt signaling and you develop the following phenotype:
- Decreased osteoblast activity and increased osteoblast apoptosis
- Increased osteoclast activity
- Decreased bone mass due to #1 and #2
- Decreased beta-islet cell mass in the pancreas and reduced insulin
- Increased gluconeogenesis by the liver
- Increased LDL cholesterol
- Increased atherosclerosis
- Increased blood pressure
All of the above phenotypes are classically seen with human aging. This is why GSK-3β inhibition alone will probably not “solve” aging. Instead, we must do something about oxidative stress as well, probably something about P66shc expression.
We expect that the next blog in this Strange But Powerful Molecules Series will be about P66shc.

Hello, Dr. Watson.
Thank you for the excellent information regarding GSK-3 A and B. I especially enjoyed the repetition of phosphorylation, ubiquitination and proteasomal degradation of catenin and various other downstream targets of GSK-3 inhibition/activation; it really hammered the idea home.
However, you seem to have used the wrong image in the degradation of beta catenin with the absence of WNT stimulation, it should be this one, I think.http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4102778/figure/F5/
On a separate note, I don’t usually ask questions that I don’t have much understanding of, but I figured that since you’ve helped me to learn so much, I could at least try.
Regarding amyloid beta plaques, tau phoshorylation, the role played by GSK-3B regarding the former two, the inhibition of GSK-3B by IFN-Y and the activation of GSK-3B by h. pylori, could it be that, similar to the counter-intuitive tumor suppressor activity displayed by GSK-3B when activated and inhibited, a similar process is playing out that induces amyloid beta placque formation and tau tangles in the presence of either viral or bacterial infection? That is, might H. pylor activation of GSK-3B be of practical equivalence, regarding amyloid plaques and tau tangles, to HSV-1-induced, IFN-Y-mediated GSK-3B inhibition? I ask because there seems to be a growing concept involving HSV-1 latent infections, recurrence during immunosenescence and AD pathology.
Herpes simplex virus type 1 and Alzheimer’s disease: increasing evidence for a major role of the virus.
http://www.ncbi.nlm.nih.gov/pubmed/25157230
It’s just a thought, and one with not enough time put behind it, but it’s given in thanks.
I look forward to learning about P66shc from you.
Pingback: Lithium for Life Extension? | Josh Mitteldorf
Pingback: h+ Magazine | Lithium for Life Extension? - h+ Magazine
Hi,
I’ve already compiled a list of some natural GSK3b inhibitors
http://selfhacked.com/2014/02/09/the-benefits-of-lithium/#Other_Natural_GSK-3b_Inhibitors
Fractals in Biology and Medicine, Volume 4. pag.257-268. 2005
edited by Gabriele A. Losa, Theo F. Nonnenmacher, Ewald R. Weibel
Evolution and Regulation of Metabolic Networks
Giuseppe Damiani
Istituto di Genetica Molecolare, Consiglio Nazionale delle Ricerche, Via Abbiategrasso 207, 27100,
Pavia, Italy; e-mail: damiani@igm.cnr.it
Summary. The analysis of metabolic processes, gene expression patterns, and protein–protein interactions in different organisms indicates that cellular metabolic networks have a scale-free and hierarchical topology described by power laws. The dynamics of these networks might be produced by a fractal organization of an autoregulatory loop, named metabolic hypercycle, between opposite redox processes of anabolic and catabolic types. This fractal architecture allows the formation of a long range correlated state of cellular networks which is globally regulated by a critical hub sensitive to the redox state. In prokaryotic cells this fundamental regulator is generally a two-component kinase system while in eukaryotic cells it is likely that casein kinase-2 and glycogen synthase kinase-3 play a central role in metabolism control. Both prokaryotes and eukaryotes share the same conserved sequence signatures, the PAS domain, in the main sensors of the changes in redox potential. Many experimental data support the hypothesis that the developmental pathways of cells and complex organisms are the results of conserved biological clocks based on metabolic hypercycles organized in fractal networks.
http://biomedphys.sgu.ru/Files/LIB/Springer/%20Fractals%20in%20Biology%20and%20Medicine/25.pdf
I can’t wait to read this and be overwhelmed by the implications. Thank you for your work.
Pingback: Литий для увеличения продолжительности жизни? | «Инфо-СМИ»
Pingback: Литий для увеличения продолжительности жизни? | 360 NEWS
Pingback: Главные новости дня » Литий для увеличения продолжительности жизни?
Pingback: Литий для увеличения продолжительности жизни? | LastPortal.Ru
Pingback: Литий для увеличения продолжительности жизни? | Новости России и мира | Сми, новости, Финансы, Происшествия, Украина
Pingback: Литий для увеличения продолжительности жизни? | 12 Новостей
Pingback: Литий для увеличения продолжительности жизни? | Woman Max
Pingback: Литий для увеличения продолжительности жизни? | Новости
Great work. Wish could read this earlier. Here is some GSK relevant inhibitors, the information listed can help to have a rough idea about the kind of substance, http://www.bocsci.com/description.asp?cas=1346704-33-3&cid=84
Saving the time to look for them in the Internet.
Pingback: ON AGING - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™
Pingback: Литий для увеличения продолжительности жизни? | sde.in.ua