
By James P Watson with editorial assistance and contributions from Vince Giuliano
I. WHY YOU SHOULD BE INTERESTED IN HUTCHINSON-GILFORD PROGERIA SYNDROME (HGPS)
Summary: HGPS is a major clue to solving the “puzzle of aging” and the molecular mechanisms here are relevant to normal aging.
HGPS is one a dozen monogenetic diseases that accelerates aging and is extremely rare, occurring in only 1 out of every 4-8 million babies born. However, the same protein that builds up in these children (called progerin) also builds up in your skin with normal aging and you can accelerate your own progerin-related aging with exposure to long wave length UV light! The mechanism that accelerates aging in HGPS and in normal skin is the same. It is called “Alternative Splicing” but in reality, there is “nothing alternative about it.” It happens with normal aging even without UVA light exposure. This is why you should be interested in the molecular mechanisms of “alternative splicing,” a mechanism discussed in the previous blog entry Transposable DNA elements – Part 3.
This disease just puts an exclamation point on how important the alternative splicing subject is for all of us, not just for children born with this terrible disease. Alternative splicing also appears to play a major role in sporadic cases of inherited and sporadic Autism Spectrum Disorder. It also plays a major role in depression and anxiety. It also plays a role in schizophrenia, cancer, arteriosclerosis, diabetes mellitus, and hundreds of extremely rare diseases. For this reason, Vince Giuliano and I have written these blog entries to help our readers understand “alternative splicing”, and have chosen HGPS as an illustration, a “case report” on how alternative splicing works.
Interestingly, we also report on very recent research about how manifestations of progerin-related aging can be reversed at least on the cell level with the aid of certain simple familiar chemicals.
Because this blog entry goes into considerable technical depth on several arcane subtopics, it is easy to lose track of what the important messages are. Here is a list of them as I (Vince) see them.
- HGPS is now a well understood model of accelerated aging. We think we know a lot about the molecular mechanisms of it and what might be done about them.
- In a nutshell, HGPS is due to a single-point mutation on a cryptic splice site in the Lamin A gene. The result is the production of progerin instead of Lamin A, a molecule with a farnesylated tail that cannot be cleaved off as normal, meaning that proteins responsible for the integrity of internal cell structures are not produced. When Lamin A does not develop and mutant prelamin A (progerin) accumulates, hetrachromin anchoring proteins disappear. As a result, the heterochromatin (compacted chromatin around silenced DNA) cannot be anchored to the nuclear envelope. Then, the histones can no longer silence the DNA that should be silenced (Ex: transposable elements, pseudogenes, micro satellite regions, etc.). Consequently, transposable elements become activated and this induces DNA damage and much additional mischief.
- HGPS is an interesting and highly informative model of ordinary aging. The changes that happen over 15 years in HGPS also normally happen in dermal cells and vascular cells of normal individuals, but at a much slower rate. We all seem to have slow-acting HGPS.
- HGPS provides an example of how alternative splicing can trigger or accelerate the entire aging cascade. It also provides an example of how a single gene abnormality can trigger the entire aging process.
- There are several known substances that seem to reverse HGPS progeria and restore cellular morphology and operations to normal, on the cell level at least. One of these, identified in 2011 is rapamycin. More-recently identified are the familiar substances methylene blue and the cabbage-broccoli phytochemical sulfaforaphane. Therapies so simple as proper mitochondrial antioxidants may hold the key to reversal of HGPS phenotypical symptoms on a cell level. As yet, however it does not appear that this knowledge has either been translated into an effective therapy for HGPS or explored as a therapy for ordinary aging.
- HGPS acts via a program of aging, following a known sequence of well-defined steps, although of course these are subject to a certain amount of stochastic variability. Comparison of HGPS and normal aging gives additional credence to the fact that normal aging is also a programmed phenomenon and that the typical period of human aging is probably not immutable.
- This blog explores many related technical topics such as morpholino oligonucleotides, cytoplasmic SRSF1and farnesylation inhibitors. And there are many complex diagrams illustrating molecular mechanisms. Please don’t let the technical detail derail you from getting these central messages.
- More important results are sure to come soon. There are too may leads and clues to cures for HGPS reported here for the research situation to slow down soon. And in the process we may well get some important additional clues as to how further slow down normal aging.
As background, Vince provided an introduction to alternative splicing and a discussion of it in the context of genomic evolution in the blog entry just preceding this one Transposable DNA elements – Part 3 TEs and and other key mechanisms of evolution: incRNAs, A to I editing, alternative splicing and exonization. Also, as a bit of ancient history, Vince published a short blog entry introducing HGPS back in 2009 Progerin, HGPS and a possible new theory of aging. The present blog entry expands on and updates that one.
II. ABOUT SAM BURNS
To put a face on this terrible disease, We would like to share the story of Sam Burns, a child who was born to normal parents and has become the poster child for HGPS. Unfortunately, Sam died last year, despite all of the efforts of modern science to ameliorate the disease. Here is his picture and his story.


Image source: Image source: Sam at the age of 14 and his mother
Meet “Sam”, a happy child who has touched the lives of millions people with his positive attitude about life, despite being diagnosed with HGPS. The picture on the right is a picture of Sam with his mother when Sam was 14 years old. He died in 2014 when he was only 17 years old. About 350 people worldwide have HGPS like Sam. All of them die by age 20. Sam went public about his condition in the HBO documentary, “Life According to Sam” and a book about him was written. See the trailer here. Despite this dreadful condition that has killed everyone afflicted with HGPS, Sam remained upbeat about life and has shared his philosophy about how to live a happy life with 30 million people via a TEDx talk. His TEDx talk is currently the 2nd most viewed TEDx talk out of over 30,000 TED talks that have been given.
Sam lived in Foxborough, Massachusetts and went to high school, like everyone else. He played percussion in the band and this was one of his favorite things to do. He would have graduated this spring. A living memorial to him is being created now by his friends and family. If he had survived, he would have graduated from high school this year. Sam’s Mother, Leslie Gordon, PhD, an Associate Professor of Pediatrics at Brown University, was instrumental in starting the Progeria Research Foundation. This foundation funded the recently published study which showed that rapamycin decreased the levels of the mutant form of Lamin A, called progerin, which builds up in the cells of HGPS patients. Rapamycin is one of three strategies described below that have been shown to help treat this disease.
Following are some references about Sam and the Progeria Research Foundation. Please view the TEDx talk that Sam Burns gave. It is very touching and true for all of us.
Ref: https://www.youtube.com/watch?v=36m1o-tM05g&app=desktop
Breaking News: Sam Berns’ TEDx Talk Reaches 10 Million Views
<iframe width=”640″ height=”360″ src=”https://www.youtube.com/embed/36m1o-tM05g” frameborder=”0″ allowfullscreen></iframe> embed code
2015 Living memorial being built as tribute to Sam Berns HBO documentary called ‘Life According to Sam’ gave people a rare look at progeria
2011 Good news in fight against premature aging disease
Despite being so rare, HGPS has received more attention than any other genetic mutation disease because it so closely “mimics” the normal picture of aging. A main difference between normal aging and HGPS is that the entire “aging program” is completed in about 15 years with HGPS, whereas with normal aging, the “aging program” takes almost 100 years to complete. Whereas a few people with normal aging can live as long as 122 years, almost all of the people with HGPS die by 16-20 years of age. The most common findings in HGPS include features that mimic normal aging, such as alopecia, skin atrophy, mottled pigmentation of the skin, generalized lipodystrophy, joint stiffness, arthritis, arteriosclerosis, coronary artery disease, left ventricular enlargement, and strokes. However, HGPS children also have unique findings that do not “mimic” normal aging, such as absent eyebrows, prominent eyes/proptosis, micrognathia, open cranial bone fontanelles, and absent sexual maturation. This is why some have called HGPS a “caricature of normal aging”, rather than a “copy of normal aging.”
For most of us, we don’t really care about this semantic difference. We are really looking for an answer to the question “what is the true cause of aging” and are hoping to find some clues in this rare, but tragic condition. First I will talk about how our skin also accumulates the same protein that accumulated in Sam’s body and how this affects our skin and blood vessel aging.
III. ALL OF US HAVE “DERMAL PROGERIA”
Summary: Long wavelength UV light (e.g. tanning booth light) induces oxidative stress, which induces alternative splicing and the use of the cryptic splice site of LMNA, which produces the mutated prelamin A protein in both young and old human skin fibroblasts! This is a major cause of skin aging and skin cancer.
Key distinctions
Before diving in further, here is a review of a few key distinctions related to alternative splicing for those of you less familiar with this area:
Alternative RNA splicing — “RNA splicing is an essential and precisely regulated post-transcriptional process that occurs prior to mRNA translation. It is thought that at least 70% of the approximately 25,000 genes in the human genome undergo alternative splicing and that, on average, a given gene gives rise to 4 alternatively spliced variants – encoding a total of 90,000 -100,000 proteins which differ in their sequence and therefore, in their activities.
A gene is first transcribed into a pre-messenger RNA (pre-mRNA), a copy of the genomic DNA containing both introns (destined normally to be removed during pre-mRNA processing) and exons (usually destined to be retained within the mRNA in order to code the protein sequence).
During RNA splicing, exons are either retained in the mRNA or targeted for removal in different combinations to create a diverse array of mRNAs from a single pre-mRNA. This process is known as alternative RNA splicing.”
Citation and following illustration source
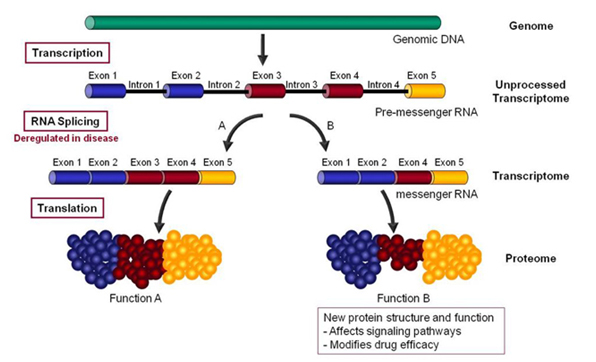
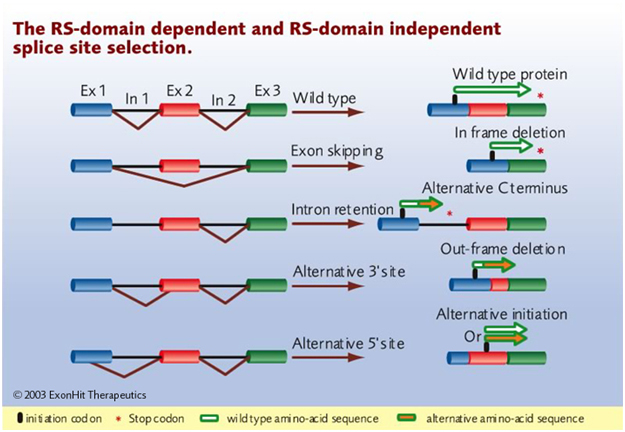
‘RNA splicing is specifically deregulated in disease conditions. A precise understanding of these deregulations can reveal new targets for the discovery of more efficacious drugs or new biomarkers for the development of more accurate diagnostics.”
Spliceosome: “Pre-mRNA splicing is an essential, precisely regulated process that occurs after gene transcription and prior to mRNA translation. Pre-mRNA splicing begins with the ordered assembly and coordinated action of the particles U1, U2, U4, U5 and U6 snRNPs (small nuclear ribonucleoprotein particles) and non-snRNP proteins on the pre-mRNA. Each snRNP particle contains a small nuclear RNA molecule (snRNA) and several proteins. The complex of snRNPs and non-snRNPs is called the spliceosome. The process of pre-mRNA splicing can be divided into three stages (Visit the diagram from the Molecular Cell Biology book for more details or the animated movie –” Source; Human Splicing Finder , pre messenger RNA splicing. You can also check out these additional explanations.
Cryptic splice site: “Eukaryotic genomes contain large numbers of splice sites, known as cryptic splice sites (css), which are generally held to be disadvantageous sites that are dormant or used only at low levels unless activated by mutation of nearby authentic or advantageous splice sites (1,2). Once activated, css may be used extremely efficiently, resulting in a wide range of genetic disease (3–5). It is generally accepted that css are suppressed by nearby stronger splice sites and that splice site selection can be viewed as a competition between the various potential splice sites in a pre-mRNA for the splicing machinery (1,2). — For genes with many introns it is suspected that up to 50% of mutations that cause disease do so by affecting splicing, either through the activation of css, exon skipping or disruption of alternative splicing (4–7). Css are found in exons as well as introns and their recognition by the splicing machinery is similar to splice site recognition in general and is dependent upon information both at the splice site and outside this region at enhancer and silencer sequences (8–10).” From the 2011 publication Cryptic splice sites and split genes.
HGPS and progerin indeed have a lot to do with normal aging
Until 2006, no one really thought that there was much true overlap between HGPS and normal aging. After all, many experts called HGPS a “caricature of aging”, saying that this disease had no resemblance to normal aging. Even expert scientists studying the biology of aging did not think that progerin, the mutant form of prelamin A, accumulated in cells undergoing normal aging. No one says that today. All of that old “dogma” has been thrown out the window as several independent research teams have published data between 2006 and 2013 which confirm that progerin accumulates in normal skin cells with aging and triggers the same cellular and molecular features of HGPS (which will be explained in detail in section #3 below). Here are the studies that have over-turned the “dogma:”
2006 Lamin A-Dependent Nuclear Defects in Human Aging. Scaffidi and Mistelli from the NIH laboratories showed that the same cryptic splice site in exon 11 of the LMNA gene is used by the spliceosome in healthy individuals and contributes to normal aging in everyone. Whereas this alternative splice site is used constitutively in HGPS, it is used only sporadically in normal aging but creates the same nuclear defects as HGPS (abnormal nuclear shape, changes in histone modifications, and increased DNA damage).
2007 – The Mutant Form of Lamin A that Causes Hutchinson-Gilford Progeria Is a Biomarker of Cellular Aging in Human Skin McClintock and a group from Columbia University, Brown University, and the NIH did a series of biopsies in normal individuals from newborn to age 97 showed that progerin mRNA and progerin protein levels were measurable in 150 skin biopsies from all age groups at low levels, but that the progerin protein was primarily localized in the skin in a subset of dermal fibroblasts near the basement membrane and the papillary dermis in young people, whereas with aging the number of dermal fibroblasts with progerin increased and extended to the deep reticular dermis in older patients. They documented that progerin was a biomarker of normal cellular aging and was linked to “terminal differentiation” and cellular senescence.
2013 – Longwave UV Light Induces the Aging-Associated Progerin Takeuchi and Runger showed that UVA light (315-400nm) but not UVB, induced progerin expression and HGPS-like changes to all cells exposed to UVA light, but more in aged cells. The molecular mechanism was UVA induction of reactive oxygen species (singlet oxygen), which triggered alternative splicing of the LMNA pre-mRNA, and the subsequent production of progerin proteins. A single dose of UVA radiation increased progerin production by 2.5-2.7 fold, but this increase did not induce levels of progerin like what was seen at baseline in skin cells derived from HGPS patients where the progerin mRNA levels were 47.5 fold higher. Moreover, the increased levels of progerin mRNA returned to normal within 24 hours in young skin fibroblasts but did not in normal skin from older patients or in skin from HGPS patients. A well designed part of this study proved that the increase in progerin mRNA was not due to an overall increase in the expression of the LMNA gene, since the normal LMNA pre-mRNA did not increase with UVA or UVB light exposure (in other words, UV light does not increase the transcription of the LMNA gene, but it changes the alternative splicing of the LMNA gene).
There was some “good news” in this landmark study by Takeuchi and Runger. They repeated the experiment in normal dermal fibroblast cells, but added an anti-oxidant to the in vitro study that quenched singlet oxygen free radicals (sodium azide). This completely inhibited the production of progerin after exposure to UVA light, but did not decrease baseline progerin in the dermal fibroblasts. This study lends credence to the idea that anti-oxidants may help prevent but not treat skin aging.
Summary: The studies above clearly show that use of the cryptic splice site in exon 11 occurs with normal aging from age 0 to 97 and this induces alternative splicing of the LMNA pre-mRNA, producing progerin proteins with normal skin aging. It also shows that UVA light (315-400 nm) accelerates this phenomena, but not UVB light (which damages DNA and RNA) which does not induce alternative splicing. Previous studies had shown that UVC also induces alternative splicing of several genes that regulate apoptosis and cell proliferation (exon 4 skipping in the PIG3 transcripts, alternative splicing of MDM2 and MDM4 transcripts, alternative splicing of ABL1, CHEK2, and MAP4K2 transcripts). Thus the “old dogma” that HGPS and normal aging have no similarities is clearly wrong and has been abandoned.
Additional References:
2009 DNA Damage Regulates Alternative Splicing through Inhibition of RNA Polymerase II Elongation
IV. WE ALL HAVE “VASCULAR PROGERIA“
Summary: The LMNA gene is alternatively spliced in our blood vessels with telomere shortening-induced cellular senescence, along with 3 other alternatively spliced genes. We all have the same kind of progeria as in HGPS. It is just working a lot slower.
A. Introduction
Recently, a publication by Olive and colleagues showed that progerin builds up in a few of the cells in adventitia of arterial walls, with progerin being present in only 1 out of every 1,000 cells in a one month-old newborn, to 20 in 1,000 cells by 97 years of age. The increase amounted to a 3.34% increase per year. Interestingly, the progerin in these cells was accumulating in the cytoplasm, which suggested that the mutant protein was accumulating over time after repeated mitotic events. When these cells were analyzed, it was clear that the same cryptic splice site in exon 11 was being used in these endothelial cells which came from normal, healthy volunteers. Here is a photomicrograph of the progerin building up in the adventitia, media, and plaque in coronary blood vessels in non-HGPS patients:
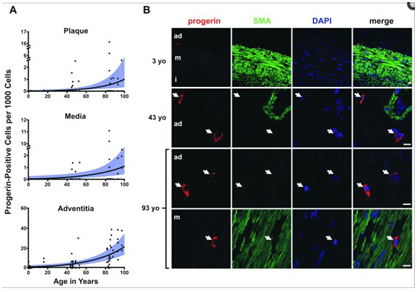
Reference: 2010 Cardiovascular Pathology in Hutchinson-Gilford Progeria: Correlation with the Vascular Pathology of Aging “…Importantly, though progerin was detected at higher rates in the HGPS coronary arteries, it was also present in non-HGPS individuals. Between ages one month and 97 years, progerin staining increased an average of 3.34% per year (P<0.0001) in coronary arteries. — Conclusion: We find concordance between many aspects of cardiovascular pathology in both HGPS and geriatric patients. HGPS generates a more prominent adventitial fibrosis than typical CVD. Vascular progerin generation in young non-HGPS individuals, which significantly increases throughout life, strongly suggests that progerin has a role in the CV aging of the general population.”
Another recent study showed that cellular senescence activates the LMNA cryptic splice side and induces progerin production, whereas immortalized cells suppressed progerin production. When they looked closely at the cause of cellular senescence, they showed that the production of progerin was dependent on telomere shortening. Specifically, this happens when cellular senescence was induced by an oncogene (H-rasV12) or by sodium butyrate, an HDAC inhibitor that induces cell senescence in cells with normal telomeres. Here is data from this:
Image reference: (2011 Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts)
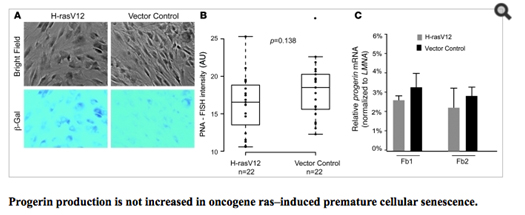
The most interesting study, however, was a recent publication that showed how alternative splicing in 4 key genes occurs with endothelial cell senescence. One of these 4 genes that undergoes senescence-associated alternative splicing is the LMNA pre-mRNA. This study is covered in detail below.
B. Alternative Splicing Plays a Role in Endothelial Senescence
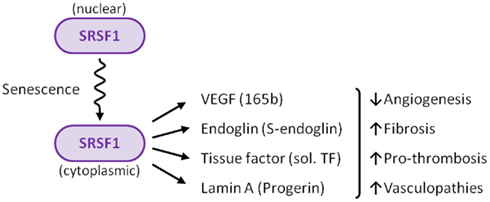
SRSF1 appears to be the regulator of how much and what kind of alternative splicing occurs in endothelial cells
The possibility that alternative splicing played a role in endothelial senescence spawned a flood of research over the past 8 years that has led to several ground-breaking discoveries. Specifically, there is now strong evidence that alternative splicing of at least 4 gene pre-mRNAs plays a major role in endothelial cell senescence, atherosclerosis, hypertension, coronary artery disease, cerebrovascular disease, renovascular disease, and peripheral artery disease. The most studied splicing factor that controls alternative splicing of genes involving atherosclerotic disease is serine-arginine splicing factor 1 (SRSF1). SRSF1 plays a role in both “constitutive splicing” and “alternative splicing” of pre-mRNA, but has many other non-spacing roles in the cell as well (mRNA nuclear export, mRNA stability, mRNA quality control, mRNA translation, maintenance of genomic stability, and oncogenic transformation.
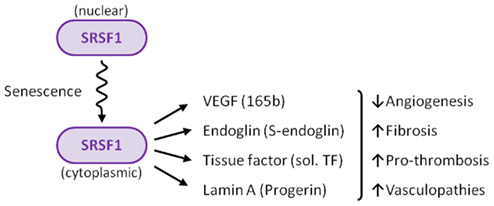
2012 The splicing factor SRSF1 as a marker for endothelial senescence (full)
C. Examples of four genes that are alternatively spliced in endothelial cells – leading to endothelial cell senescence, atherosclerosis, CAD, hypertension, stroke, etc.
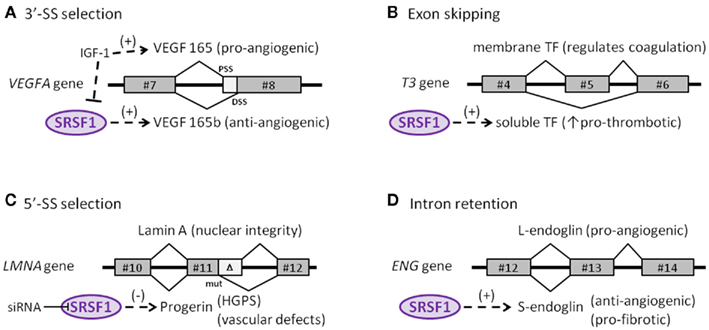
Although more alternatively spliced genes may be found that play a role in endothelial cell senescence, now it is clear that at least four genes undergo alternative splicing that are regulated by SRSF1. This includes vascular endothelial growth factor A (VEGFA), tissue factor (TF) gene (T3), the lamin A gene (LMNA), and the endoglobin gene (ENG). Interestingly, each of these four genes undergo a different type of alternative splicing. For this reason, this is a good time to delve into the different types of alternative splicing that have been discovered.
C1. Tissue Factor gene (T3) – An example of “exon skipping” in a key endothelial gene involving cell senescence
The protein made by tissue factor gene (T3) is the primary initiator of blood coagulation. With atherosclerosis, a state of hypercoaguability exists that is due in part to the Tissue Factor protein (TF). Normally, TF is expressed as a transmembrane glycoprotein and is not secreted into the bloodstream, but when tissue trauma occurs or when there are pro-inflammatory cytokines in the bloodstream, a soluble form of TF is produced and secreted by endothelial cells. This soluble form of the TF protein occurs because of alternative splicing that is due to the activation of SRSF1. When SRSF1 is activated or over-expressed, exon #5 is “skipped” during pre-mRNA splicing. Exon #4 is directly connected to exon #6, producing an mRNA with a “translational frameshift”, leading to the production of a soluble form of TF. The “exon skipping” type of alternative splicing of the T3 gene is illustrated below:
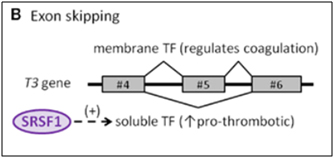
Image source: 2012 The splicing factor SRSF1 as a marker for endothelial senescence
One of the consequences of circulating pro-inflammatory cytokines is deep venous thrombosis (DVT) and arterial clots with heart attacks and strokes. The pro-inflammatory cytokine induced alternative splicing, directed by SRSF1, leads to exon #5 skipping of the T3 gene, thereby creating a soluble, secreted form of Tissue Factor (TF). This phenomenon contributes to endothelial senescence and the disease called arteriosclerosis.
C2. Vascular Endothelial Growth Factor A gene (VEGFA) – An example of abnormal 3’ splice site (3’-SS) selection in a key endothelial gene involving cell senescence.
The VEGF gene is a well-known growth factor gene that is normally thought of as having only an angiogenic role. However, this is a major misconception! The VEGFA gene actually can be either pro-angiogenic or anti-angiogenic, depending on how it is spliced. The VEGFA gene actually produces two families of isoforms that are generated by alternative splice sites in its terminal exon #8. In response to IGF-1 signaling, SRSF1 is inhibited and the spliceosome chooses the 3’ proximal splice site (PSS) in exon 8, which produces a pro-angiogenic isoform of VEGF (aka VEGFa). However if TGF-beta signaling occurs and overpowers IGF-1 signaling, SRSF1 is activated and then chooses the 3’ distal splice site (DSS) in exon 8, which produces an anti-angiogenic isoform of VEGF, called VEGFb. Here is a diagram illustrating two alternative splice sites in the VEGF pre-mRNA:
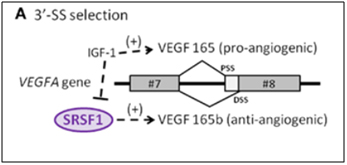
Image source: 2012 The splicing factor SRSF1 as a marker for endothelial senescence
Conclusion about VEGFA: IGF-1 induces the use of the proximal 3’ splice site in exon #8 and inhibits SRSF1, producing a vascular endothelial growth factor isoform that is pro-angiogenic. TGF-beta extinguishes this effect activates SRSF1, which chooses the distal 3’ splice site in exon #8, producing a vascular endothelial growth factor isoform that is anti-angiogenic. The phenomena occurs with endothelial cell senescence.
C3. Endoglin gene (ENG) – An example of intron retention in a key endothelial cell gene involving cell senescence — explains TGF-beta mediated vascular fibrosis, endothelial telomerase suppression, and varicose veins!
Endoglin is an auxiliary co-receptor for TGF-beta that is found on the surface of endothelial cells. It plays a key role in vascular remodeling and angiogenesis. Mutations in this gene lead to a terrible disease called “hereditary hemorrhagic telangiectasia Type I.” The phenotype of this disease is vascular malformations. When endothelial cells are not senescent, they express a pro-angiogenic long isoform of the gene called “L-endoglin”. However, when endothelial cells become senescent, they express an alternatively spliced version of the protein called “S-endoglin” which is anti-angiogenic and pro-fibrotic. The difference between the L-endoglin and S-endoglin pre-mRNA transcripts involves the inclusion of the intron between exon #13 and #14. If this intron is excluded, you get L-endoglin and the protein product is pro-angiogenic. If this intron is included, due to SRSF1 activation of the spliceosome, then you get S-endoglin and a protein product that produces fibrosis of the blood vessel wall. The details on how this works is fascinating, since it explains why the telomerase gene (hTERT) is down-regulated in endothelial cells, in response to TGF-beta/Smad3/c-myc signaling.
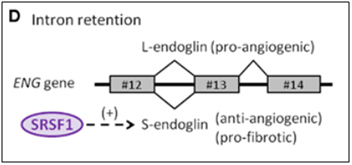
Image source: 2012 The splicing factor SRSF1 as a marker for endothelial senescence
This S-endoglin alternative-splicing induced effect also explains why varicose veins occur, also due to TGF-beta signaling. However, the details of how this works is complicated and is beyond this introduction to alternative splicing. For more information, however, look up some of the references below.
References:
2007 TGF-beta1 upregulation in the aging varicose vein
2007 Mechanisms of action of TGF-beta in cancer: evidence for Smad3 as a repressor of the hTERT gene
2002 The helix-loop-helix protein id-1 delays onset of replicative senescence in human endothelial cells
Conclusion about ENG: The endoglobin gene plays a key role in endothelial cells, since it encodes an auxiliary receptor for TGF-beta, a growth factor involved with fibrosis, endothelial senescence, varicose veins, and telomere shortening in endothelial cells. The ENG gene transcripts undergoes alternative splicing, producing two proteins with almost opposite effects, depending on SRSF1 splicing factor activation. When SRSF1 is not activated, the ENG pre-mRNA undergoes normal splicing. When SRSF1 is activated, intron inclusion occurs of the intron between exon #13 and #14, producing a protein that favors TGF-beta/Smad4/c-myc signaling. This pathway leads to fibrosis of the blood vessel wall, the down regulation of the hTERT gene in endothelial cells, and the pathology of varicose veins. This all starts with the inclusion of one intron!
C4. Lamin A gene (LMNA) – Example of alternative 5’ splice site (5’-SS) selection in a key endothelial cell gene involving cell senescence
The Lamin A gene is ubiquitously expressed in all mammalian cells, since it is an indispensable structural member of the nuclear envelope. As was seen in HGPS, in normal skin fibroblast aging, and in UVA-induced skin fibroblast aging, the same cryptic splice site in exon #11 is also used in endothelial cell aging (senescence). Here SRSF1 triggers the use of the cryptic splice site in response to oxidative stress (ROS). This produces a pre-mRNA that is 150 nucleotides short and a prelamin A protein that is missing 50 amino acids. This shorter version of prelamin A does not go through the last of 4 steps in post translational modification of normal prelamin A, leading to a mutant form of prelamin A that cannot be incorporated into the nuclear envelope. As a result, the mutant prelamin A builds up in the cell and leads to endothelial cell senescence. Here is an illustration of this abnormal 5’ splice site selection in the LMNA gene.
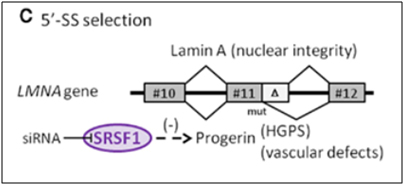
Image reference:2012 The splicing factor SRSF1 as a marker for endothelial senescence (full)
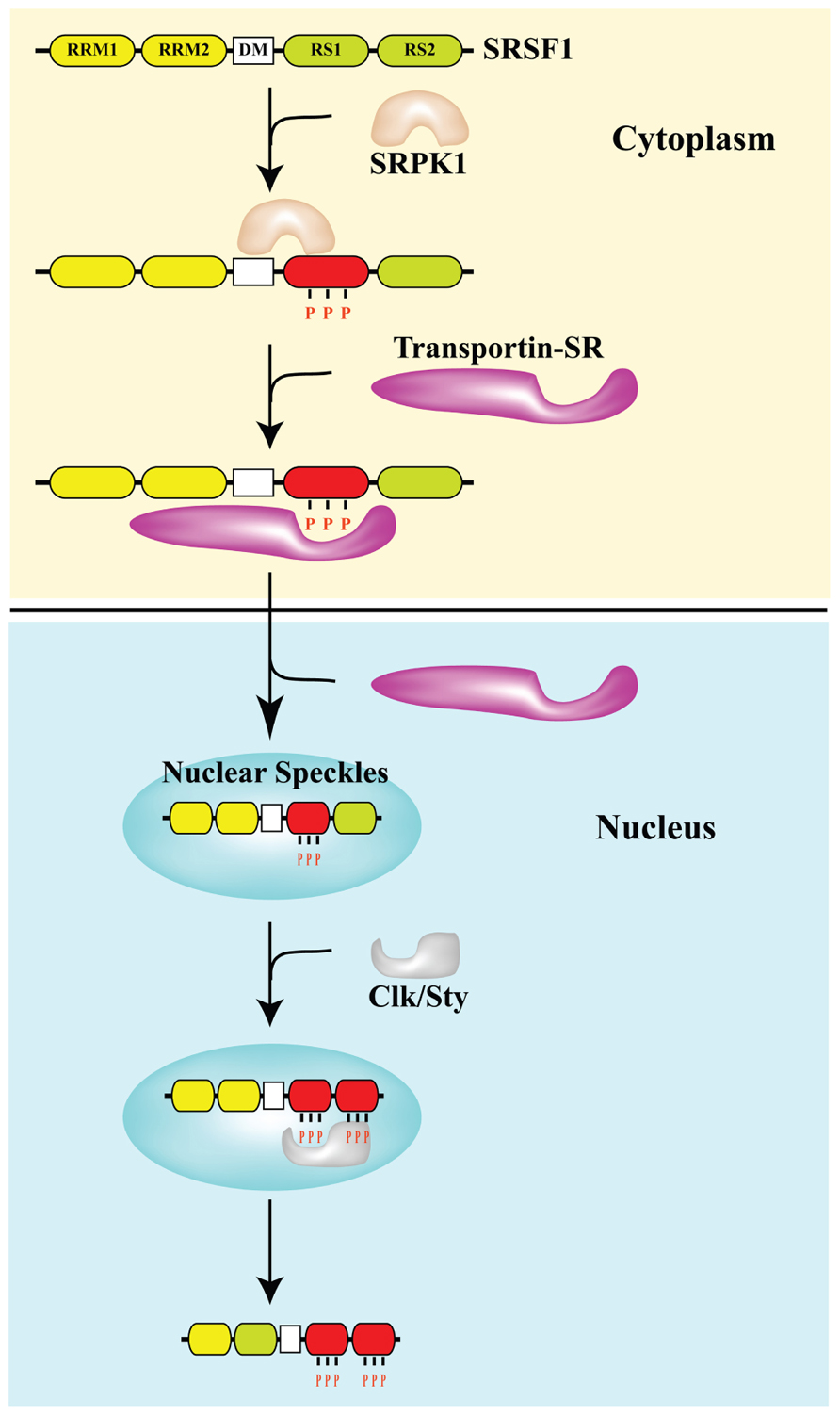
D. What triggers SRSF1-induced alternative splicing? – Cellular senescence and cellular stress induces cytoplasmic SRSF1-mediated alternative splicing. As a consequence, it is actually the accumulation of SRSF1 in the cytoplasm that occurs with cellular senescence.
D1. The many “jobs” of SRSF1? – SRSF1 has 5 “jobs” in the nucleus and 2 “jobs” in the cytoplasm.
By now we hope you are convinced that alternative splicing is an important subject. We have raised more questions than we have answered. However, it should also be clear by now that SRSF1 is a key “player” in the alternative splicing game. Some would say that SRSF1 is the “card dealer” that is mixing up the “exon cards” in this game of “gene cards”. To call SRSF1 a “splicing factor”, however, is a gross understatement! SRSF1 has been shown to have many different functions, depending on where it is located. In the cell nucleus, it controls normal alternative splicing, miRNA processing, nonsense-mediated RNA decay, and mRNP export. In the cytoplasm, it activates mTOR, which enhances mRNA translation initiation. Another function of it in the cytoplasm is to mediate splicing of mRNA in the minor spliceosome, found in the cytoplasm. Here is a diagram of 6 of these 7 “jobs” that SRSF1 has:
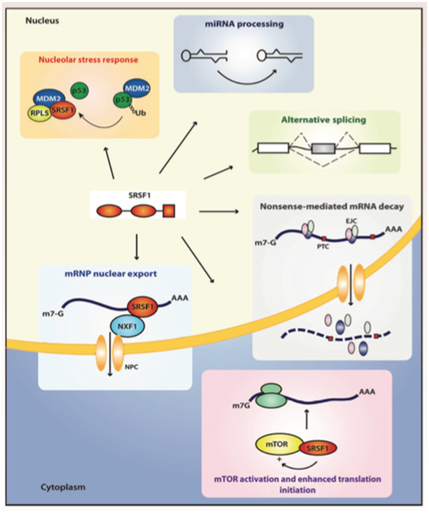
Image reference: 2014 Emerging Functions of SRSF1, Splicing Factor and Oncoprotein, in RNA Metabolism and Cancer “Figure 2. The multifunctional SR protein SRSF1. Splicing-dependent and independent functions of SRSF1 in the nucleus and the cytoplasm. EJC, exon junction complex; NFX1, nuclear exchange factor 1; NPC, nuclear pore complex; PTC, premature termination codon.”
Here is a list of the protein interactions of SRSF1 and its binding partners:
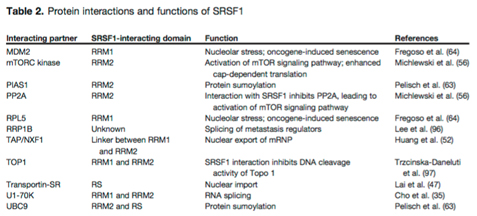
Table reference: 2014 Emerging Functions of SRSF1, Splicing Factor and Oncoprotein, in RNA Metabolism and Cancer
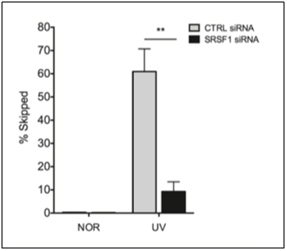
By now, we are sure that you are curious as to what activates SRSF1? The simple answer is that it depends on if SRSF1 is doing “constitutive splicing” or “stress-induced splicing”. SRSF1 is necessary for both types of splicing events, so it is not necessary for cellular stress to occur for SRSF1-mediated constitutive splicing events to occur. With alternative splicing, however, the usual triggers are stresses. Not only oxidative stress, but other stressors can also activate SRSF1, such as oncogenic stress, UV-induced stress, X-ray induced stress, and osmotic stress. However, SRSF1 must undergo phosphorylation by another splicing factor before both constitutive and alternative splicing can occur. There are several enzymes that phosphorylate SRSF1 at specific sites, including Clk/Sty 1,2,3, and 4 kinases; SRPK 1 and 2 kinases; and topoisomerase I. SRSF1 is also dephosphorylated by phosphatases 1 and 2 if SRPK1 or 2 phosphorylates SRSF1 at a specific site that induces nuclear transport of SRSF1. The actual transport occurs by a transport protein called “Transportin-SR”. Clk/Sty kinases further phosphorylate SRSF1 in the nucleus. This moves SRSF1 from the nuclear speckles to the active sites of transcription, where it promotes splicing. Below is a graph of the effect of SRSF1 in exon skipping of the MDM2 pre-mRNA exon 11, which occurs when MCF-7 cells were radiated with UV light, with and without siRNA knockdown of SRSF1.
References:
2015 The RNA Splicing Response to DNA Damage
2015 Splicing factor SRSF1 negatively regulates alternative splicing of MDM2 under damage
D2. The Key to Understanding SRSF1: Location, Location, Location!
What is more important than the source of cellular stress is the location of SRSF1. As they say in real estate, the 3 keys are “location, location, location”. The same is true for SRSF1. Normally, SRSF1 is found in both the cytoplasm and in the nucleus. It can phosphorylate the major spliceosome proteins in the nucleus (triggering normal splicing of pre-mRNA) or it can remain in the cytoplasm where it can phosphorylate the minor spliceosome proteins in the cytoplasm (triggering alternative splicing of mRNA). What determines if SRSF1 stays in the cytoplasm or migrates into the cell nucleus? The protein kinases SRPK1 and Clk/Sty determine this. SRPK1 is another important splicing factor that must be present in the cytoplasm to phosphorylate SRSF1 in response to cellular stress. If the “dose” of stress is not too large, then SRPK1 stays in the cytoplasm and continuously phosphorylates SRSF1. Once SRSF1 is phosphorylated by SRPK1, then a transport protein called “Transportin-SR” can bind to SRSF1 and “carry” it into the cell nucleus. When SRSF1 migrates into the cell nucleus, it localizes to heterochromatin granules called “nuclear speckles”. Thus the phosphorylation of SRSF1 by SRPK1 allows SRSF1 to migrate into the cell nucleus and participate in “constitutive splicing” events.
Thus the “location, location, location” rule for normal splicing is the nuclear localization of SRSF1, where SRSF1 mediates normal splicing. This is illustrated in the diagram below:
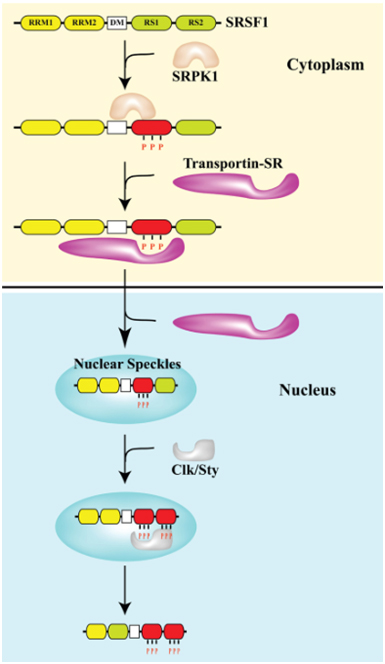
Image reference from 2013 Pre-mRNA processing factors meet the DNA damage response
D3. What happens with high dose stress? – Cytoplasmic “Location, Location, Location” of SRSF1
When the dose of cellular stress is high enough (due to ROS, UV, X-rays, osmotic stress, heat, etc.), it can trigger the check-point pathway mediated by p53 and Rb. This triggers cellular senescence. (A thorough discussion of cellular senescence is beyond the scope of this blog). When this occurs, SRPK1 is released from its binding partners (heat shock proteins and co-chaperones) and is phosphorylated by Akt. This triggers the nuclear localization of SRPK1. When this occurs, SRPK1 can no longer phosphorylate SRSF1, and as a result, SRSF1 cannot migrate to the nucleus. As a consequence, more SRSF1 is left behind in the cytoplasm. This sounds “paradoxical”, but this is actually what occurs. SRSF1 can actually modulate alternative splicing in the cytoplasm of some mRNA, such as the ENG mRNA, since this involves the minor spliceosome.
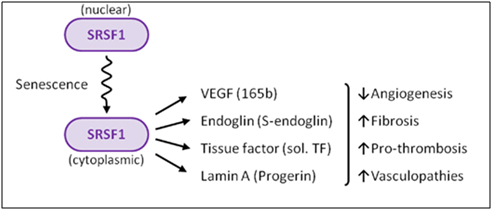
Image reference: 2012 The splicing factor SRSF1 as a marker for endothelial senescence (full)
E. What other factors determine Exon Skipping vs Exon Inclusion – UV light and Topoisomerase inhibitors acting on RNA Polymerase II
Cellular stressors such as UV light and genotoxic stress also can affect alternative splicing via other molecular mechanisms besides SRSF1 and SRPK1. One example of this is how UV light or topoisomerase I inhibitors like camptothecin affect exon skipping or exon inclusion. UV light or camptothecin both slowed the rate at which RNA Polymerase II elongation occurred, as it copied the RNA transcript from DNA. When RNA Polymerase II was transcribing at a fast rate, exon skipping would occur sometimes. When UV light or camptothecin were administered to colon cancer or breast cancer cells in vitro, exon skipping did not occur. The details of exactly how this occurs is too complicated to present here. Here is a reference for those who wish to delve into this in more detail.
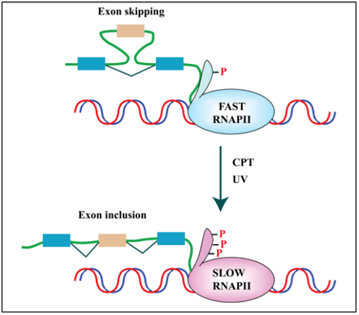
Reference and image source: 2013 Pre-mRNA processing factors meet the DNA damage response. “FIGURE 2. DNA damage affects splicing decisions by modulating the phosphorylation status of RNAPII and the elongation rate of transcription. CPT-induced Top1ccs have immediate and specific effects on RNAPII. CPT triggers a high degree of phosphorylation of the largest subunit (Rpb1) of RNAPII (Baranello et al., 2010). Ultraviolet (UV) irradiation affects cotranscriptional alternative splicing in a p53-independent manner, through hyperphosphorylation of the RNAPII carboxy-terminal domain (CTD) and subsequent inhibition of transcriptional elongation (Muñoz et al., 2009).”
Summary: In this section, we clearly showed the scientific evidence that progerin accumulates as a function of chronological aging in the blood vessel walls of normal individuals who do not have HGPS. Although the adventitia has the highest concentration of progerin, it also accumulates in the media and intima as well. Progerin accumulation in the walls of blood vessels only affects 1 out of every 1,000 cells at birth and increases at a rate of 3.34% per year. As the endothelial cells divide, they “pass on” the progerin to subsequent generations of daughter cells. Cellular senescence induced by short telomeres but not by oncogenes is the necessary trigger for the activation of the cryptic splice site in the LMNA exon 11. At least 3 other genes undergo alternative splicing with endothelial cell senescence – VEGFA, ENG, and T3. These 3 genes and LMNA are each alternatively spliced by different splicing methods, with each one undergoing the following alternative splicing:
| Gene | Location of SRSF1 & AS Method | Trigger of Alternative Splicing | Consequence of AS |
| T3 | cytoplasmic SRSF1-induced exon skipping of exon #5 | Pro-inflammatory cytokines or cellular senescence | Tissue factor protein becomes prothrombotic and soluble (circulates in plasma) |
| VEGFA | cytoplasmic SRSF1-induced distal 3’ splice site in exon #8 | TGF-beta cytokine or cellular senescence | VEGF becomes anti-angiogenic |
| ENG | cytoplasmic SRSF1-induced intron inclusion | cellular senescence | Endoglin protein acts as a TGF-beta receptor, inducing fibrosis, down regulation of hTERT, etc. |
| LMNA | cytoplasmic SRSF1-induced 5’ splice site in exon #11 | oxidative stress or replicative senescence | prelamin A induces aging of the cells, loss of heterochromatin mark,
H3K9me3, loss of HP1a |
These four genes are all induced by the cytoplasmic localization of SRSF1, a splicing protein that is involved in a diverse set of cellular activities including alternative splicing, miRNA processing, RNP export out of the nucleus, nucleolar stress response, nonsense-mediated mRNA decay in the nucleus, and the activation of mTOR in the cytoplasm. In summary, SRSF1 plays a key role in alternative splicing and incellular senescence.
V. HGPS: AN EXAMPLE OF HOW ALTERNATIVE SPLICING CAN TRIGGER AND ACCELERATE THE ENTIRE AGING CASCADE
Summary:The trigger of aging in HGPS is a single mutant protein that cannot get rid of its farnesylated “tail” (no “clipping site” on the protein). The “unclipped”, farnesylated version of prelamin A accumulates and causes accelerated aging.
The first obvious lesson we can learn from HGPS is that “a single gene abnormality can trigger the entire aging program”. This is not to say that aging is simply due to a cryptic space site mutation in the LMNA gene, but rather that one cryptic splice site can trigger all of aging. HGPS is not the only disease that can do this. Several other diseases due to single point mutations in completely different genes can do the same thing (Ex: Werner’s syndrome, Cockayne syndrome, xeroderma pigmentosum, Fanconi’s anemia, etc.). However none of these other mutations produce an “aging phenotype” at such a young age or that has such a close resemblance to normal aging. For instance, Werner’s syndrome does produce an accelerated aging phenotype that looks like normal aging, but occurs later in life (This is why Werner’s syndrome is often called “adult progeria”). In the case of HGPS, the aging trigger is a point mutation in a exon for a protein-coding gene called called Lamin A (LMNA). LMNA codes for a key structural component of the nuclear cytoskeleton, which is required to maintain nuclear shape and DNA organization. Maintaining this 3D structure of the nucleus is vital for normal cellular function. Lamin A, Lamin B, and Lamin C produce a nuclear scaffold along with several other Lamin proteins, which creates the 3D structure of the nucleus. In HGPS, the point mutation in the LMNA gene causes an abnormal splice signal in codon 608 in exon 11 of the LMNA pre-mRNA. At this point, a cytosine is substituted for a thymine nucleotide (C > 608 T). Here is an illustration of this:
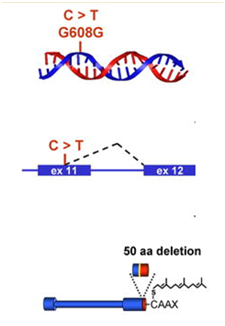
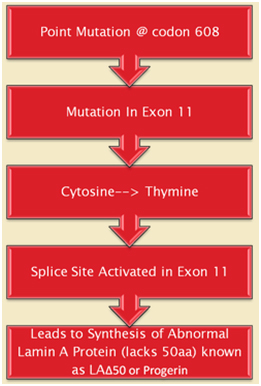
Illustration source: What causes progeria?
This results in part of exon 11 being excised with the adjacent intron (between exon 11 and exon 12). As a result, the last 150 amino acids of the prelamin A protein are missing. When the last 150 nucleotides of exon 11 are cut off by the spliceosome, the mRNA produces a mutant pre-protein called prelamin A, with 50 missing amino acids. This prelamin A does not get processed correctly during post-translational modification. Here are the post translational modifications of the prelamin A protein in wild type and HGPS prelamin A:
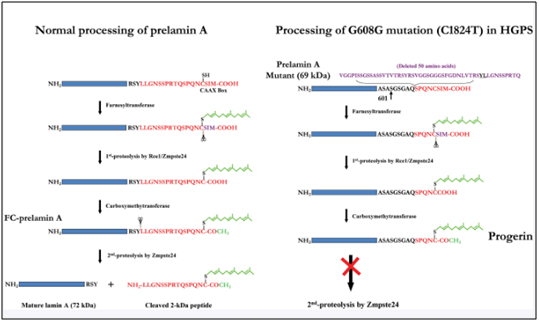
Image source: 2009 Genomic Instability and DNA Damage Responses in Progeria Arising from Defective Maturation of Prelamin A
When prelamin A is not processed correctly and does not loose its “tail, the protein is called “progerin”. It does not get incorporated into the nuclear envelope correctly and accumulates in the nucleus. As a result the Lamin Associated Domains (LADs) of condensed chromatin do not get anchored to the nuclear envelope. Here is an illustration of how the lamin proteins are so important for anchoring condensed chromatin (silenced DNA) to the nuclear envelope.
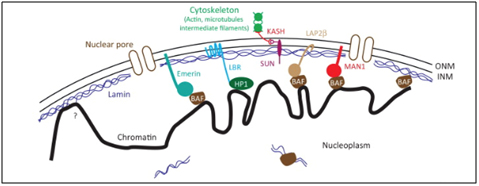
Image reference: 2013 Mechanisms of heterochromatin subnuclear localization
When lamin A does not develop and mutant prelamin A (progerin) accumulates the anchoring proteins disappear (like HP1a). As a result, the heterochromatin (compacted chromatin around silenced DNA) cannot be anchored to the nuclear envelope. Heterochromatin is normally localized in this subnuclear location.
As a result, the histones can no longer silence the DNA that should be silenced (Ex: transposable elements, pseudogenes, micro satellite regions, etc.). Consequently, the transposable elements become activated and this induces DNA damage. The following illustration shows two chromosomes (blue and green) and how their condensed chromatin (heterochromatin) is anchored to the lamina of the nuclear envelope.
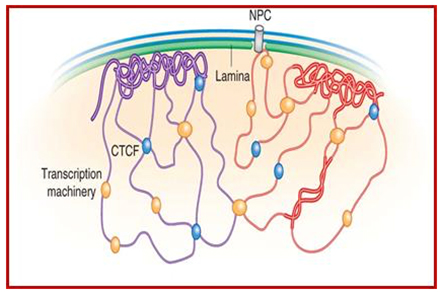
Image and legend source: Chromosomal Nature-Before, During and After Gene Activation:
“Lamin Associated Domains LADs may consist of relatively condensed chromatin (thick lines) and aggregate at the nuclear lamina. Other repressed regions may interact with each other in the nuclear interior, as do active regions. Complexes formed by components of the transcription machinery (transcription factories) and CTCF may tether active regions together. Parts of only two chromosomes are depicted, each in a different color for clarity. Most interactions occur within chromosomes, and relatively few occur between chromosomes;Bas van Steensel & Job Dekker; http://www.nature.com/”
Summary: 85-90% of HGPS cases are due to a single point mutation in codon 608 in exon 11 in the LMNA gene that encodes for two proteins – Lamin A and Lamin C. As a result, a cryptic splice site is used to “excise” the last 150 nucleotides of axon 11 during the processing of the LMNA pre-mRNA. This produces a pre protein that is 50 amino acids too short and does not contain the “endoproteolytic site” that is used by the enzyme Zmpste24 to “cut the tail” of the prelamin A. As a result, prelamin A cannot be incorporated into the cell nuclear cytoskeleton and the following cascade of events happens. It is this cascade of events that “causes the aging”, not the prelamin A, per se.
- Loss of the normal size and shape of the cell nucleus (Lamin A is a nuclear structural protein) in 70% of the cells. The nuclei have wrinkles, herniations, and lobules of their nuclear element.
- Loss of other nuclear cytoskeleton proteins – With HPGS, not only does Lamin A levels decrease by 95% in the cell nucleus, there is also a 6-fold decrease in the amount of Lamin B in the nucleus as well. Lamin B is an important nuclear cytoskeleton protein that is encoded by the same gene (LMNA. In HGPS, there is a 70% decline in Lamin B levels too. Other nuclear cytoskeletal proteins are also depleted or misplaced, such as the lamina-associated polypeptides (LAP2s), which are also depleted 6-fold.
- Loss of chromatin anchoring to the nuclear cytoskeleton. With HGPS, there is a 2-fold decrease in the protein (HP1), which is an anchoring protein that tethers chromatin (histone proteins) to the nuclear cytoskeleton (i.e. lamina proteins). HP1 is called an “adaptor protein” or “linker protein” that secures the chromatin to the nuclear skeleton, allowing the chromatin to be organized in a 3D fashion.
- Reduction or complete loss of “junk DNA” silencing. There is a very important histone mark that indicates the DNA wound around the nucleosome is “turned off” or “silent”. This histone mark is the trimethylation of lysine 9 on histone subunit H3 (aka H3K9me3). With HGPS, in 60% of the cells the H3K9me3 histone modification is lost. H3K9 trimethylation (H3K9me3) is a universal feature of heterochromatin, the part of our DNA that should remain “silenced”. When histone H3K9 trimethylation is lost, the DNA is no longer compacted and the “junk DNA” gets transcribed. This is a bad thing that leads to the re-expression of transposable elements, pseudogenes, and satellite repeats, which should NOT be transcribed. Thus the loss of HP1 and H3K9me3 that occurs as a result of the abnormal lamin A-induced loss of 3D structure of the nucleus means that a massive amount of “junk DNA” gets transcribed, which is a major molecular feature of aging.
VI. THERE IS HOPE FOR HGPS – THE AGING PROGRAM CAN BE REVERSED OR SLOWED DOWN
Another important lesson we can learn from HGPS is that this disease shows that aging is more like a program which is accelerated from the normal 100 years (normal aging) to 15 years (HGPS). Nature has given us many examples of “molecular programs”, such as gestation (9 months long), embryogenesis (12 weeks long), apoptosis (3-5 days long), circadian rhythms (1 day long), and ultradian rhythms (less than 24 hours long). It is hard for most people to accept that aging is programmed too, but there is good evidence for this. The best line of evidence against a non-programmed type of aging is that 100% of humans develop the same set of features with aging in a similar order of events (presbyopia, alopecia, whitening of the hair, skin thinning, atherosclerosis, skin discoloration, menopause, andropause, osteoporosis, arthritis, cognitive changes, sarcopenia, etc.) If aging was truly a random, stochastic event, the features of aging would occur in random order and would not affect 100% of human beings (i.e. random events don’t happen 100% of the time in the same order). The fact that over 80% of the features of normal aging are also seen in children with HGPS suggest that this disease is truly an accelerated aging model. The million dollar question, however, is can this aging program be reversed or slowed?
The answer for HGPS is “yes” and the three methods below illustrate how this can be done, at least on the cellular level.. The first method tackles the “upstream cause” of HGPS. The next two tackle more of the “downstream effects” of HPGS. All three showed amazing results and have huge implications for human aging. And, more recent even more amazing methods are reported in subsequent sections below.
- Morpholino oligonucleotides – In 2005, the first effective in vitro therapy for progeria cells derived from 5 individuals who actually had HGPS was demonstrated by Paola Scaffidi and Tom Mistily at the National Cancer Institute, NIH (ref). Although their proof-of-concept experiment was only a test tube study, they were able to reverse the cellular and molecular features of HGPS cells, restoring normal ellipsoid shape of the cells and restoring the production of normal, non-mutant lamin A proteins. Surprisingly, the strategy also restored normal lamin B, LAP2, and HP1⍺ in the 90% of the cells. Even the H3K9me3 histone status was restored. Reversal of the HGPS phenotype on a cellular and molecular level did not require the cells to divide (mitosis) and did not require the cells to disassemble their nuclear cytoskeleton (which occurs with mitosis). What was most amazing was that normal gene expression levels also occurred in the 5 genes that they checked (MMP3, HAS3, TIMP3, MMP14, and CCL8). These fibroblast genes are dysregulated in both HGPS and normal aging (ref). How did they induce such an astounding reversal of such a horrible disease? By blocking the abnormal splice side on the LMNA pre-mRNA. This was accomplished with a synthetic strand of nucleotides called a “morpholino oligonucleotide”.
The morpholinos had a sequence of nucleotides that perfectly matched the 25 nucleotide region at the abnormal splice site in exon 11 of the LMNA pre-mRNA. (i.e. it was an antisense strand of nucleotides). Morpholinos are not made of normal DNA or RNA and therefore do not work (transiently) like siRNA do to block processed mRNA. Instead, morpholinos are synthetic sequence that look like RNA or DNA, but do not have ribose and phosphate groups like RNA or DNA. Instead, the ribose (or deoxyribose) sugars have been replaced with 6-membered “morpholino rings” and the phosphate linkers have been replaced with hosphorodiamidate groups. Here is picture of a morpholino oligonucleotide and an illustration of of a morpholino binding to the alternative splice site in an exon of a pre-mRNA (not exon 11 in HGPS).
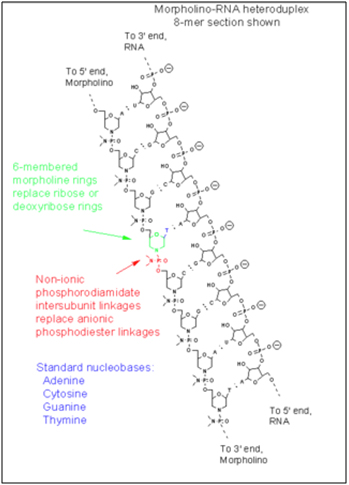
Image source: Wikipedia Morpholino
As a result of the two synthetic changes to the oligonucleotide, the morpholinos are very stable and resist degradation by morpholine rings. They are very stable and cannot be degraded by enzymes (Nucleases) that degrade nucleic acid fragments. Scaffidi and Mistily took cultured fibroblast cells derived from patients with HGPS and showed that an antisense morpholino that covered up the a cryptic splice site on the pre-mRNAs transcribed from the LMNA gene. The morpholinos blocked the pre-mRNA before the splicing machinery could get to the pre-mRNA. Then when the splicosome removed the introns from the LMNA pre-mRNA, they did not excise the last 150 oligonucleotides from exon 11, thereby allowing a full length Lamin A protein to be made in the ribosomes.
- Farnesyl Transferease Inhibitors – Lamin A, like most proteins, undergoes post translational modifications. Before the Lamin A protein is modified, it is called “prelamin A”. Prelamin A undergoes four, sequential post translational modifications, as follows:
- Farnesylation – Prelamin A has 98 amino acids on its C-terminus that are not present in the final processed lamin A protein. This 98 amino acid “tail” of prelamin A has a CaaX motif that triggers an enzyme called “Farnesyl Transferase” to add a 15 carbon isoprenoid lipid to the protein called “farnesyl”. Here is a diagram of this farnesyl attached to the protein:
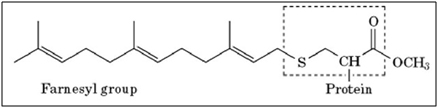
Image source: What-when-how — Farnesylation
Additional reference: 2006 Prelamin A Farnesylation and Progeroid Syndromes
B. Endoproteolytic release of 3 amino acids – The next step in post-translational processing of prelamin A is the removal of three amino acids from the “tail” of prelamin A. This is probably done by the enzyme, Zmpste24 (see illustration below).
C. Methylation of the carboxyterminus – The 3rd step in the post translational processing of prelamin A is the methylation of the “tail” of prelamin A. This is probably done by the enzyme, Icmt. This step is also illustrated below. Unfortunately, in HGPS, this prelamin A is a mutant protein and does not undergo the last step in post translational processing, which is to remove this farnesylated, methylated “tail” of prelamin A.
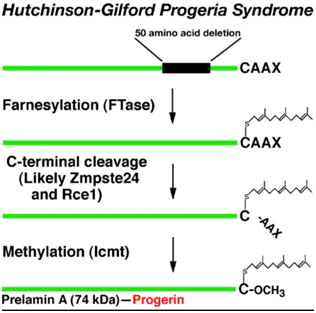
D. Clevage of the farnesylated, methylated “tail” of prelamin A – All of the above post-translational modifications of prelamin A must then be removed before prelamin A becomes “lamin A”. Interestingly, the same enzyme that carries out step #2 also carried out this step Zmpste24, a zinc metalloproteinase enzyme. Here is a diagram illustrating the difference between normal prelamin A processing and HGPS prelamin A processing, where this step does not occur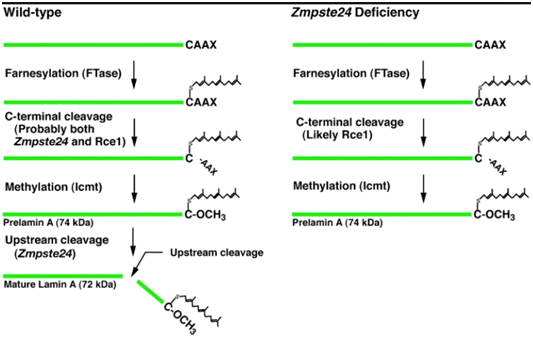
Additional reference: 2004 Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice
Inhibition of the first step in the above process has been shown to alleviate some of the symptoms of HGPS in cell models of the disease, in animal models of the disease, and in human clinical trials of the Farnesyl Transferase inhibitors. Farnesyl Transferase inhibitors are of great interest in cancer biology, since they showed a lot of in vitro promise in treating cancers with mutations in the ras family of oncogenes (H-ras, N-ras, and K-ras). The proteins produced from each of these genes undergo farnesylation, just like prelamin A does. As a consequence, farnesyl transferase inhibitors have shown promise in clinical trials for cancers where these genes are muted, such as pancreatic cancer (80% have K-ras mutations), colon cancer (40-50% have K-ras mutations), thyroid cancer (50-80%) have mutations in either K-ras, N-ras, or H-ras), and many other cancers. Here are some structures of different farnesyl transferase inhibitor drugs that have been developed for cancer.
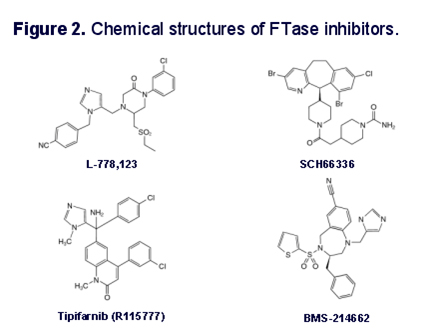
Image reference: 2004 Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice
Unfortunately, the clinical trials of farnesyl transferase inhibitors (FTIs) for cancer have all failed and none of been FDA approved to date. This has been due mostly to side effects of the drugs, since they not only inhibit the farnesylation of ras pre-proteins, but also inhibit the farnesylation of many other pre-proteins during post translational modification. For HGPS, only one FTI has completed phase II clinical trials. In progeria patients, this FTI, called “lanafarnib”, reduced the prevalence of stroke, transient ischemic attacks, and headaches. It also improved height and diminished the rigidity of the bones in these progeria patients when combined with a cocktail of two other drugs.
References:
2012 Experimental Drug Is First To Help Kids With Premature-Aging Disease
- mTOR inhibitors – In 2012, Cao and colleagues from the NIH laboratory reported on a remarkable in vitro experiment where they treated HGPS fibroblasts with the mTOR inhibitor, rapamycin. Remarkably, rapamycin abolished nuclear blabbing, delayed the onset of cellular senescence, and enhanced the degradation of the progerin proteins in the HGPS fibroblasts. Rapamycin also induced autophagy, which decreased the insoluble progerin aggregates.
References:
Here is a diagram from an editorial written about this subject in a simple, clear cut mechanistic explanation:
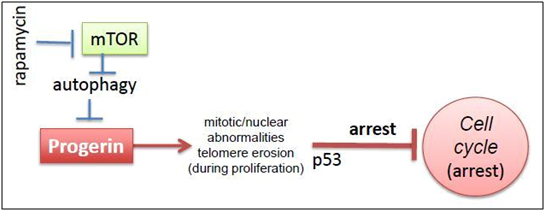
2011 Image source: Progeria, rapamycin and normal aging: recent breakthrough
VII. Why the HGPS de novo mutations are only heterozygous (single copy) and why the inherited HGPS mutations require two copies homozygous) to create symptoms.
Understanding the molecular biology behind the principles of “loss of heterozygosity” and “autosomal dominance”
HGPS has one very odd finding. Some cases require two “bad copies” of the LMNA gene (14%), whereas the majority of cases have only one “bad copy” of the LMNA gene (86%). When it takes two bad copies to produce the disease and the gene is found on a non-sex chromosome (i.e. not an X or Y), this is called “homozygous autosomal recessive”. When the disease can be manifest even if only one copy of the gene is mutated (even though there is a normal copy of the gene), this is called “heterozygous autosomal dominant”.
Another odd finding in HGPS is that all of the homozygous autosomal recessive cases are due to inherited “bad copies” of the LMNA gene. This means that both parents have to have a “bad copy” of the LMNA gene and then must pass this “bad copy” on to their children. However, the other odd finding in HGPS is that all of the heterozygous autosomal dominant cases are due to de novo mutations, which means the single “bad copy” of the LMNA gene occurred spontaneously, in the germ line cell from the mother’s ovary or father’s testes. No cases of heterozygous autosomal dominant HGPS have been found, since these children cannot have children of their own (they are sterile and die as teenagers).
Reference: 2016 Homozygous missense mutation in the lamin A/C gene causes autosomal recessive Hutchinson-Gilford progeria syndrome
How can we understand this “heterozygous” vs “homozygous” paradox in HGPS? Well, we have to go back and look at what portion of the LMNA gene is mutated to understand why certain parts of the gene produce symptoms with only one copy (autosomal dominance) whereas other parts of the gene produce symptoms only if both copies mutated (autosomal recessive). Without understanding this, the puzzle will never be solved and we will miss a critical principle that is key to understanding aging and cancer – the concept of “loss of heterozygosity”.
a. Mutations in non-splice sites create HGPS only if both copies are bad (i.e. heterozygous autosomal recessive). There are other mutations in the LMNA gene that produce HGPS. One of these is the “K542N” mutation, found in exon 10. This mutation is called a “missense mutation” since it involves a protein coding part of the gene and it changes the amino acid sequence of the lamin proteins A and C from a lysine to an arginine. The “K542N” mutation can be passed on from generation to generation because carrying one copy of the gene does not cause the disease. If a person only has one copy of the “K542N” mutation in the LMNA gene, they do not develop HGPS and are phenotypically normal. If the person (inherits) two copies of the “K542N” mutation, they develop the disease. When geneticists looked at the gene sequence mutated in the “K542N” cases, they found that this mutation DID NOT affect splicing! It was NOT in a splice site. Instead, it was a coding portion of the gene (exon 10) shared by both the lamin A and lamin C versions of the protein (i.e. both splice variants, but not the splice site). This discovery was a huge breakthrough in understanding why and how both copies of the gene had to be mutated if it was a “non-splice site” mutation, whereas with the splice site mutation, only one copy of the gene had to be mutated.
Reference: 2004 Homozygous missense mutation in the lamin A/C gene causes autosomal recessive Hutchinson-Gilford progeria syndrome
b. Splice-site mutations create HGPS only if one copy of the gene is bad (i.e. homozygous autosomal dominant). The mutations are point mutations in a the LMN gene, found on the 1st chromosome, on the long arm at region 22 (1q22). 86% of the LMN mutations involve a heterozygous mutation of a splicing site, called the “G608G mutation”, which is expressed in an autosomal dominant manner. These heterozygous autosomal dominant cases are all de novo mutations and are therefore not inherited (since the carriers die before they can have children). However, autosomal recessive modes of inheritance have been found and are due to homozygous mutations in gremlin cells. Here the mutation is called the “K542N mutation”. With the K542N mutation, heterozygous carriers are phenotypically normal and this is why the mutation can be passed on from generation to generation.
VIII. AN OLD FAVORITE CHEMICAL -METHYLENE BLUE – APPEARS TO COMPLETELY RESCUE PROGERIN-AFFETED HGPS CELLS AND COULD POSSIBLY BE USEFUL FOR TREATING HGPS ITSELF
The research supporting this finding was published in Aging Cell with issue date of Dec 14 2015 in the publicatiom Methylene blue alleviates nuclear and mitochondrial abnormalities in progeria “Hutchinson–Gilford progeria syndrome (HGPS), a fatal premature aging disease, is caused by a single-nucleotide mutation in the LMNA gene. Previous reports have focused on nuclear phenotypes in HGPS cells, yet the potential contribution of the mitochondria, a key player in normal aging, remains unclear. Using high-resolution microscopy analysis, we demonstrated a significantly increased fraction of swollen and fragmented mitochondria and a marked reduction in mitochondrial mobility in HGPS fibroblast cells. Notably, the expression of PGC-1α, a central regulator of mitochondrial biogenesis, was inhibited by progerin. To rescue mitochondrial defects, we treated HGPS cells with a mitochondrial-targeting antioxidant methylene blue (MB). Our analysis indicated that MB treatment not only alleviated the mitochondrial defects but also rescued the hallmark nuclear abnormalities in HGPS cells. Additional analysis suggested that MB treatment released progerin from the nuclear membrane, rescued perinuclear heterochromatin loss and corrected misregulated gene expression in HGPS cells. Together, these results demonstrate a role of mitochondrial dysfunction in developing the premature aging phenotypes in HGPS cells and suggest MB as a promising therapeutic approach for HGPS.”
The article notes a number of other interesting observtions related to HGPS not covered so far in this blog entry, I (Vince) thought this passage on to be particularly interesting: “Progerin suppresses the expression of PGC-1α PGC-1α serves as a master inducer of mitochondrial biogenesis through its co-activation of nuclear respiratory factors (NRFs), which control the expression of nuclear genes encoding mitochondrial proteins (Wu et al., 1999; Finck & Kelly, 2006). Using adipogenesis array, we previously reported that in HGPS adipocytes, PGC-1α was the most severely downregulated gene among the 84 genes involved in energy metabolism (Xiong et al., 2013). Thus, to understand how progerin causes mitochondrial defects, we first examined PGC-1α expression in HGPS fibroblasts. Quantitative RT–PCR experiments revealed that the PGC-1α mRNA level declined by eight folds in HGPS cell lines compared to normal cell lines (Fig. 3A). Immunofluorescence with an anti-PGC-1α antibody showed weakened or a complete loss of PGC-1α nuclear staining in HGPS cells (Fig. 3B). Western blotting analysis further confirmed the reduction of PGC-1α protein in HGPS cells (Fig. 3C). Consistent with the downregulation of PGC-1α, we found that most of PGC-1α’s downstream target genes, including Nrf1, Tfam1, Mfn1, Mfn2, Opa1, Fis1, and Drp1 (Dillon et al., 2012), were significantly suppressed in HGPS cells (Fig. 3D). We further confirmed the inhibitory effect of progerin on PGC-1α using lentiviruses expressing GFP-progerin (Fig. 3E). To test whether restoration of PGC-1α in HGPS cells alleviates the mitochondrial defects, lentiviruses carrying either a control lamin A gene (LA) or human PGC-1α gene were applied to HGPS fibroblast cells (Fig. 3F). To our surprise, we did not detect any obvious improvements in mitochondrial morphology or behavior (representative cell images shown in Fig. 3G). Instead, an elevated mitochondrial ROS in PGC-1α-expressing cells was found, in comparison with the control lamin A (LA)-expressing cells (Fig. 3F). Taken together, these results suggest that while PGC-1α has an established role in regulating mitochondrial biogenesis, correction of PGC-1α expression alone might not be sufficient to rescue the severe mitochondrial phenotypes caused by progerin.”
IX. SULFORAPHANE ALSO APPEARS TO ENHANCE PROGERIN CLEARANCE
A 2015 article in Aging Cell. Reports on how another familiar substance appears to be able to clear progerin from HGPS cells and reverse phenotypic HGPS symptoms on a cell level. Sulforaphane enhances progerin clearance in Hutchinson-Gilford progeria fibroblasts. “Hutchinson-Gilford progeria syndrome (HGPS, OMIM 176670) is a rare multisystem childhood premature aging disorder linked to mutations in the LMNA gene. The most common HGPS mutation is found at position G608G within exon 11 of the LMNA gene. This mutation results in the deletion of 50 amino acids at the carboxyl-terminal tail of prelamin A, and the truncated protein is called progerin. Progerin only undergoes a subset of the normal post-translational modifications and remains permanently farnesylated. Several attempts to rescue the normal cellular phenotype with farnesyltransferase inhibitors (FTIs) and other compounds have resulted in partial cellular recovery. Using proteomics, we report here that progerin induces changes in the composition of the HGPS nuclear proteome, including alterations to several components of the protein degradation pathways. Consequently, proteasome activity and autophagy are impaired in HGPS cells. To restore protein clearance in HGPS cells, we treated HGPS cultures with sulforaphane (SFN), an antioxidant derived from cruciferous vegetables. We determined that SFN stimulates proteasome activity and autophagy in normal and HGPS fibroblast cultures. Specifically, SFN enhances progerin clearance by autophagy and reverses the phenotypic changes that are the hallmarks of HGPS. Therefore, SFN is a promising therapeutic avenue for children with HGPS.
————————————————
As a final comment, based on these very-recent results, I (Vince) would bet that there are several other common mitochondrial antioxidants that can reverse phenotypic characteristics of HGPS on a cell level at least. I am optimistic that we will soon be able to defeat this dreadful disease for once and for all. Learning about HGPS about a dozen years ago was a major emotional factor for my making longevity sciences my current career.

{kind=link}
{kind=link}
{kind=link}
Thanks for this very interesting if very demanding post. Lets assume that aging is stochastically programmed — think salmon. Further, lets assume that progerin is the agent of that programming that causes aging. Then, it would be very interesting to determine the amount of progerin with age in different species with their different lifespans. Does progerin accumulate more rapidly in dogs than in humans? More unlikely, but is there some kind of avalanche of progerin in salmon? Cross species comparisons should tell us a lot.
James and Vince,
I am a 50 year old physician with a Lamin a defect. It seems to be denovo. All siblings, aunts and uncles have been tested. Both my parents have passed, but we were able to test tissue from my mother that was from a specimen taken from her cancer before she passed. I was unable to test my father.
I have complete heart block at rest, and have been told that I am high risk for sudden cardiac death, and progressive heart failure.
After much self research, I am self medicating with rapamycin on a 6 week on, 6 week off dosing regimen in order to prevent further progression of my disease while minimizing side effects from the rapamycin.(Expensive, that rapamycin is, and of course, insurance will not cover it in this situation.)
I would like to know, specifically, which compounds you feel might help ameliorate this condition.
I am not asking for medical advice, as that would be inappropriate, but rather your opinion on which substances might hypothetically have the most benefit.
I have two young children, a great wife, and want to live as long and with as good health as possible, any advice would be appreciated.
Thank you for your website,
Your postings and insights have been extremely valuable, and I have been reading them for some time.
Tim
Pingback: Firewalls Com | UN News | Global perspective, human stories