
Written by Vince Giuliano, with major contributions by James P Watson
This is the first of a series of blog entries examining the roles of mitochondria in key biological activities. The goal of the series is to shed light on whether focusing on mitochondria can lead to new approaches for averting or curing neurological, cardiac and other diseases, and for practical interventions that extend lifespans. In my treatise ANTI-AGING FIREWALLS – THE SCIENCE AND TECHNOLOGY OF LONGEVITY originally drafted in May 2008, I discussed Mitochondrial Damage as the third-most important of some 18 theories of aging. I have also written blog entries related to mitochondrial biogenesis(ref)(ref) and to miotohormesis(ref). These have all remained highly relevant.
My recent refocusing on mitochondria is due to two factors: 1. the incredible life-extension results in rats reported in the previous blog entry Buckyballs, health and longevity – state of knowledge thought to be due to a mitochondrial antioxidant intervention, and 2. an incredibly creative series of phone and e-mail exchanges over the ten days with James P Watson (Jim). Many of the bolder ideas that will put forward in this series are Jim’s. And a number of the diagrams that appear in this series were put together by Jim. When I use “we” here, I think I am speaking for both Jim and I.
Our purpose in this and following blog entries in this series is to delve more deeply into exactly what is known and unknown and see what answers can be found to such questions as:
- Just how important is mitochondrial signaling in driving disease and aging processes that may play out and manifest through DNA genetic and epigenetic processes? Have we been looking in the right places in our search for basic causes of neurological diseases?
- In our focus on stem cells, regenerative medicine, iPSCs and cellular senescence, have we paid enough attention to mitochondrial DNA? In ignoring mitochondrial DNA senescence, are we perhaps missing where basic action is?
- Is there a solid theoretical basis for seeing a mitochondrial origin to neurological diseases like Parkinson and Alzheimer’s diseases and ALS? If so, what needs to be done to further explore new therapeutic possibilities? Further, what practical interventions might be available right now?
- Is there a solid theoretical basis for believing that mitochondrial interventions, antioxidant or otherwise, might lead to extension of lifespans? If so, what is that evidence?
- To what extent has lifespan extension through mitochondrial interventions in lifeforms actually been demonstrated? What about experiments in yeast, nematodes, mice and rats? How does this possibility as applied to humans require further testing?
- Are mitochondria subject to replicative or stress-related senescence? If so what are the key implications of that? Can such senescence be reversed?
- What roles do mitochondria play in cell senescence; can a mitochondrial intervention affect the senescence process or lead to apoptosis in senescent cells?
- What about the roles mitochondria play in various key processes and pathways such as the unfolded protein response, apoptosis in normal and cancer cells, autophagy, etc.?
- What about human antioxidant supplementation? Does it make sense given what is known about mitohormesis? When might it be particularly useful or harmful? What are its limits likely to be? Does the body change and adapt to increasing doses of antioxidants/
- Could strategies for reducing mitochondrial electron chain leakage be better ones for health and longevity than focusing on mitochondrial-active antioxidants? Do buckyballs affect electron chain leakage?
- What experimental work is needed to resolve outstanding questions? What new biomarkers are needed?
To answer these and related questions, we draw on research from many different perspectives, in many cases on work done by researchers who do not appear to be in communication with each other. These blog entries will blog will summarize information from several independent streams of publications, collectively a large number of them. Entire schools of researchers are studying closely related processes but seem to either not know of or to ignore each other’s work. Trying to put together the pieces of the jigsaw puzzle of health, longevity and aging, sometimes we wonder if all the pieces are from the same puzzle. We nonetheless try to fit those pieces together, and in doing so hope that in doing that we will be able to surface new important insights and perspectives. In some cases we speculate on how results from different streams are related, our own efforts to contribute. It is not at all an easy process and some of our conjectures could be off-base, but the process is a very exciting one for us.
This Part 1 blog reviews basic facts about mitochondria, discusses the electron transport chain and electron leakage in mitochondria. We discuss the sroles of mitochondria in aging and genetic mutational diseases for important insights they might lead us to. And we review and expand upon some of the material covered in previous blogs related to mitochondrial biogenesis and mitohormesis. Some of this material is basic and well known and some is derived from fairly new research. The next blog entry in this series, Mitochondria Part 2: mitochondrial pathways, diseases and aging will dig deeper and examine underlying pathway mechanisms for observed disease and aging effects. We will touch upon mitochondrial signaling and the mitochondrial UPR (unfolded protein response), into the roles of mitochondria in UPR in the endoplasmic reticulum, some of the pathways between mitochondria and the nucleus, and into signaling transport mechanisms such as microtubules and motor proteins. And we will speculate on a possible underlying mitochondrial cause common to aging and age-related neurological, cardiac and other diseases. As I now see it, a Part 3 blog entry will focus on practical interventions. It will review what is known about the impact of administration of key mitochondrial antioxidants, alpha-lipoic acid, acetyl-l-carnitine, melatonin and buckyballs in particular. And it will speculate on how research needs to be directed to answer critical outstanding questions.
Basics about Mitochondria
Those of you who already know about mitochondria might want to skip this section. First, as to the relevance of mitochondria from my treatise: “Mitochondria are tiny organelles within cells responsible for producing the cell’s source of chemical energy known as adenosine triphosphate (ATP). Many researchers believe that decline in the integrity of mitochondrial DNA is a root cause of aging and that maintaining mitochondrial health is central for longevity. Mitochondria, have their own DNA which is extremely subject to mutation such as resulting from attack by ROS. At least 40 diseases have been identified that involve pathology of mitochondrial functioning. They can affect brain and other nerve cells, kidneys, eyes, ears, the pancreas, and the liver. Also, damage to mitochondrial DNA can come about through insufficiency of certain micronutrients like zink and copper, and deficiencies of Vitamin D, C, E, B12, B6, niacin, and folic acid(ref). See also the candidate theory of aging based on micronutrient triage. — Cells have mechanisms for repair of damaged DNA and mitochondrial DNA, but these have their own side effects. There is a DNA repair enzyme known as NADH. However, repair results in depletion of NADH and of ATP. ATP depletion is believed to be one of the most critical factors leading to necrosis or cell apoptosis. –Mitochondria play additional critical roles in the cell life and reproductive cycle including roles involving cell signaling, growth, differentiation and death. They can be the sources of many important signals relating to cancers, neurological diseases such as Parkinson’s, cardiovascular diseases, type 2 diabetes, and optical neurological problems. Presence of oxidative conditions in a cell can signal the mitochondria to generate signals to other parts of the cell leading to cell apoptosis. It is thought that selective inhibition of this chain could be one means of retarding aging. The signaling relationships between mitochondrial DNA and regular cellular DNA is one example of the systems relationships between the various theories of aging. And it points out how addressing one “cause” of aging can sometimes also mitigate several others as well. The blog entry Mitohormesis discusses a number of processes that go on in mitochondria and the complex pathways through which they play their key role in energy metabolism.”

Mitochondria are like simple separate biological organisms, and are thought to have possibly evolved from such very long ago. In animal cells, they are in a complex set of symbiotic relationships with nuclear and other elements of the cells, relationships critical to the survival of mitochondria, the cells, the organs concerned, and the animals themselves. We believe that exploring the nature of those relationships may lead us to new insights related to diseases and aging – and may also suggest basic new health and longevity-promoting interventions.
Another good place to look for basics is of course Wikipedia: “In cell biology, a mitochondrion (plural mitochondria) is a membrane-enclosed organelle found in most eukaryotic cells.[1] These organelles range from 0.5 to 1.0 micrometer (μm) in diameter. Mitochondria are sometimes described as “cellular power plants” because they generate most of the cell’s supply of adenosine triphosphate (ATP), used as a source of chemical energy.[2] In addition to supplying cellular energy, mitochondria are involved in other tasks such as signaling, cellular differentiation, cell death, as well as the control of the cell cycle and cell growth.[3] Mitochondria have been implicated in several human diseases, including mitochondrial disorders[4] and cardiac dysfunction,[5] and may play a role in the aging process. The word mitochondrion comes from the Greek μίτος mitos, thread, + χονδρίον chondrion, granule. — Several characteristics make mitochondria unique. The number of mitochondria in a cell varies widely by organism and tissue type. Many cells have only a single mitochondrion, whereas others can contain several thousand mitochondria.[6][7] The organelle is composed of compartments that carry out specialized functions. These compartments or regions include the outer membrane, the intermembrane space, the inner membrane, and the cristae and matrix. Mitochondrial proteins vary depending on the tissue and the species. In humans, 615 distinct types of proteins have been identified from cardiac mitochondria,[8] whereas in Murinae (rats), 940 proteins encoded by distinct genes have been reported.[9] The mitochondrial proteome is thought to be dynamically regulated.[10] Although most of a cell’s DNA is contained in the cell nucleus, the mitochondrion has its own independent genome. Further, its DNA shows substantial similarity to bacterial genomes.[11] ”
.svg/400px-Animal_mitochondrion_diagram_en_(edit).svg.png)
About mitochondrial DNA
Mitochondrial DNA is very different than nuclear DNA: it is much simpler, it is organized differently, it generates far fewer proteins, and it is passed on from generation to generation differently. But like nuclear DNA it is subject to damage and mutations, Again quoting selectively from Wikipedia, I have highlighted some key points in bold italics: “Mitochondrial DNA (mtDNA or mDNA[2]) is the DNA located in organelles called mitochondria, — Mitochondrial DNA can be regarded as the smallest chromosome. Human mitochondrial DNA was the first significant part of the human genome to be sequenced. In most species, including humans, mtDNA is inherited solely from the mother[3]. — Nuclear and mitochondrial DNA are thought to be of separate evolutionary origin, with the mtDNA being derived from the circular genomes of the bacteria that were engulfed by the early ancestors of today’s eukaryotic cells. This theory is called the endosymbiotic theory. Each mitochondrion is estimated to contain 2-10 mtDNA copies.[4] In the cells of extant organisms, the vast majority of the proteins present in the mitochondria (numbering approximately 1500 different types in mammals) are coded for by nuclear DNA, but the genes for some of them, if not most, are thought to have originally been of bacterial origin, having since been transferred to the eukaryotic nucleus during evolution. — In most multicellular organisms, mtDNA is inherited from the mother (maternally inherited). Mechanisms for this include simple dilution (an egg contains 100,000 to 1,000,000 mtDNA molecules, whereas a sperm contains only 100 to 1000), degradation of sperm mtDNA in the fertilized egg, and, at least in a few organisms, failure of sperm mtDNA to enter the egg. Whatever the mechanism, this single parent (uniparental) pattern of mtDNA inheritance is found in most animals, most plants and in fungi as well. – “
“In sexual reproduction, mitochondria are normally inherited exclusively from the mother. The mitochondria in mammalian sperm are usually destroyed by the egg cell after fertilization. Also, most mitochondria are present at the base of the sperm’s tail, which is used for propelling the sperm cells. Sometimes the tail is lost during fertilization. In 1999 it was reported that paternal sperm mitochondria (containing mtDNA) are marked with ubiquitin to select them for later destruction inside the embryo.[5] Some in vitro fertilization techniques, particularly injecting a sperm into an oocyte, may interfere with this.”— Because mtDNA is not highly conserved and has a rapid mutation rate, it is useful for studying the evolutionary relationships – phylogeny – of organisms. Biologists can determine and then compare mtDNA sequences among different species and use the comparisons to build an evolutionary tree for the species examined(ref).”

Mitochondrial DNA – circular and closed in shape
A few key points are worth emphasis here, points we will be coming back to these later(ref):
- Any mtDNA an individual has is passed on only from his or her mother.
- mtDNA as in does not undergo recombination as in sexual reproduction.
- Changes to mtDNA come only from mutations, ones occuring during cell division or resulting from ROS damage.
- Mutations that occur in the mtDNA control region tend not to be repaired, since that region does not code for any specific product . Such mtDNA mutations may accumulate with aging.
- Thus, only mutations that occur in female germline cells (those that become eggs) are passed on to offspring.
Mitochondrial electron transport chain, electron leakage and free radical production
In mitochondria, energy production depends on a well-known electron transport chain. As described in Wikipedia: “Most eukaryotic cells have mitochondria, which produce ATP from products of the citric acid cycle, fatty acid oxidation, and amino acid oxidation. At the mitochondrial inner membrane, electrons from NADH and succinate pass through the electron transport chain to oxygen, which is reduced to water. The electron transport chain comprises an enzymatic series of electron donors and acceptors. Each electron donor passes electrons to a more electronegative acceptor, which in turn donates these electrons to another acceptor, a process that continues down the series until electrons are passed to oxygen, the most electronegative and terminal electron acceptor in the chain. Passage of electrons between donor and acceptor releases energy, which is used to generate a proton gradient across the mitochondrial membrane by actively “pumping” protons into the intermembrane space, producing a thermodynamic state that has the potential to do work. The entire process is called oxidative phosphorylation, since ADP is phosphorylated to ATP using the energy of hydrogen oxidation in many steps”
About mitochondrial uncoupling proteins
Mitochondrial uncoupling proteins play an important role in the electron transport chain. From The Biology of Mitochondrial Uncoupling Proteins (2004): “Uncoupling proteins (UCPs) are mitochondrial transporters present in the inner membrane of mitochondria. They are found in all mammals and in plants. They belong to the family of anion mitochondrial carriers including adenine nucleotide transporters. The term “uncoupling protein” was originally used for UCP1, which is uniquely present in mitochondria of brown adipocytes, the thermogenic cells that maintain body temperature in small rodents. In these cells, UCP1 acts as a proton carrier activated by free fatty acids and creates a shunt between complexes of the respiratory chain and ATP synthase. Activation of UCP1 enhances respiration, and the uncoupling process results in a futile cycle and dissipation of oxidation energy as heat. UCP2 is ubiquitous and highly expressed in the lymphoid system, macrophages, and pancreatic islets. UCP3 is mainly expressed in skeletal muscles. In comparison to the established uncoupling and thermogenic activities of UCP1, UCP2 and UCP3 appear to be involved in the limitation of free radical levels in cells rather than in physiological uncoupling and thermogenesis. Moreover, UCP2 is a regulator of insulin secretion and UCP3 is involved in fatty acid metabolism.”
Electron leakage from the electron transport chain in mitochondria normally produces a low level of superoxide free radicals. These are useful signaling molecules performing useful positive effects.
“A small percentage of electrons do not complete the whole series and instead directly leak to oxygen, resulting in the formation of the free-radical superoxide, a highly reactive molecule that contributes to oxidative stress and has been implicated in a number of diseases and aging(ref).” Mitochondrial electron leakage has been studied for a long time. You can go back to the 1993 publication Electron leakage from the mitochondrial NADPH-adrenodoxin reductase-adrenodoxin-P450scc (cholesterol side chain cleavage) system. You can also see (2010) Mitochondrial proton and electron leaks.
The normal “electron leak rate” (ELR). from healthy mitochondria is 2%. These electrons that leak out function to create hormetic dose signaling ROS and RNS that have many healthy effects, including the altering of redox sensitive signaling proteins such as Keap-1 and many others. ROS and RNS sensitive “redox proteins” allow for many gene pathways to be upregulated for xenobiotic enzymes and phase II enzymes that protect us from cellular stress. These pathways work well for coping with bursts of increases free radicals and peroxynitrates that occur with acute stresses such as infection, trauma, exercise, etc. See the blog entry Mitohormesis for details on this.
Regarding mitochondrial ROS generation control
It is a genetic/epigenetically controlled phenomenon, depicted out in this diagram:


![]()

Image source: James P Watson. As Jim explains it: “Representation of the major signalling pathways regulating mitochondrial ROS. p53, FOXOs and c-Myc orchestrate the transcription of genes encoding effector molecules (i.e. p66shc and MnSOD) which directly regulate mitochondrial ROS level in response to several intra- and extra-cellular stimuli. Note that p66shc acts both as a transducer and as an effector for mitochondrial oxidative stress.” From the 2009 publication The p53–p66shc–Manganese Superoxide Dismutase (MnSOD) network: A mitochondrial intrigue to generate reactive oxygen species: “Once considered as a mere by-product of respiration, mitochondrial generation of reactive oxygen species (ROS) has recently emerged as a genetically controlled phenomenon, involved in complex intracellular signal transduction cascades that directly regulate cell survival and death in responses to environmental stressors. These cascades are involved in the pathogenesis of several major age-related diseases, such as cancer and neurodegeneration, and also appear to somehow regulate the “normal” ageing process. — The present short review summarizes recent discoveries on mitochondrial reactive oxygen species regulation by p53, a tumor suppressor protein and p66shc, a protein implicated in the life-span determination. It also outlines the emerging network whereby these molecules cross-talk with each other and with the mitochondrial antioxidant system, namely MnSOD (SOD2), another life-span determining protein, to regulate oxidative stress in the organelle. This molecular circuit, which comprises two genetic determinants of longevity and a major tumor suppressor gene, also provides a theoretical framework connecting senescence and cancer.”
Extrinsic vs Instrinsic Control of ROS Production


![]()
Source: James P Watson. As Jim explains it: “Caloric restriction, the IGF-1/insulin pathway, and oxidative stress are known inducers of mitochondrial ROS production. The downstream signaling pathways that mediate this cellular response are closely interrelated. Indeed, SIRT1, activated by caloric restriction, deacetylates and thus activates the FoxO transcription factors, whose phosphorylation is controlled by the IGF-1 pathway. Furthermore, the adaptor protein p66shc, which directly regulates mitochondrial ROS production, inhibits FoxO activation.” Reference: Mitochondrial longevity pathways, Biochimica et Biophysica Acta (BBA) – Molecular Cell Research, Volume 1813, Issue 1, January 2011, Pages 260–268Interplay between the signaling pathways controlling mitochondrial ROS production.”
Now, with this base of information we go on to effects of aging and certain diseases on mitochondria, electron leakage, and ROS production.
There seems to be evidence that lifespans of species are correlated with lower rates of leakage in electron transport chains. Cutting down on mitochondrial electron leakage might be a better strategy for creating mitochondrial health than focusing on mitochondrial antioxidants.
The 2006 publication Plugging the Mitochondrial Leak reports: “– While an inverse correlation between resting metabolic rate and longevity in animals generally holds true, there are some exceptions to the rule. Birds, bats, and humans live several times longer than their metabolic rates would suggest. The reason lies in the rate at which reactive oxygen species (ROS) leak out of the mitochondrial respiratory chain, the succession of membrane-bound proteins that passes electrons from NADH to oxygen. According to Gustavo Barja at the Complutense University in Madrid, pigeons leak barely a tenth the ROS of rats, and live nearly ten times longer, yet their resting metabolic rates are similar. “ROS leakage is so low in pigeons that they can afford to have much lower antioxidant levels than rats, and still live longer,” says Barja. “The question is, why are pigeon mitochondria so leak-proof?” — The answer could have profound implications. According to Alan Wright at Edinburgh University, the cellular threshold for apoptosis is calibrated by the rate of ROS leakage: “Species that leak ROS slowly have a lower rate of apoptotic cell loss in degenerative conditions, including those that apparently have nothing to do with oxidative or nitrosative stress.” Analyzing single mutations in 10 different degenerative conditions across five species, Wright and collaborators found that age of onset and severity of disease correlates closely with the rate of ROS leakage. “If we could slow ROS leakage, there’s a prospect we could delay the onset of a wide spectrum of degenerative diseases,” he says. — In June 2005, Douglas Wallace’s group at the University of California, Irvine, showed that the approach could work in mammals.1 They generated transgenic mice that overexpress the antioxidant enzyme catalase in mitochondria (to break down hydrogen peroxide). Not only are average and maximal lifespans increased by about five months, but also degenerative conditions such as cardiac pathology and cataract formation are delayed. Other work suggests that antioxidants targeted to the mitochondria, such as mitoQ, concentrate 1000-fold in the mitochondrial matrix, where they inhibit apoptosis. But antioxidants have the potential to interfere with ROS signaling, which plays a major role in the physiology of the cell. Birds solve the problem by cutting leakage from complex I, not by raising intramitochondrial antioxidant levels. — “The critical factor determining ROS leakage is not antioxidant status but the redox state of complex I, which is the major source of ROS,” says Martin Brand at the MRC Dunn Unit in Cambridge, UK. “Redox state is dependent on numerous factors like substrate supply, ATP use, uncoupling, amount of complexes, and allosteric influences, such as Ca2+ activation or NO inhibition of cytochrome oxidase. So predicting the outcome depends on knowing the state of all these variables.”
The writer goes on: “–Such variables explain conundrums such as the exercise paradox-why physically active people don’t die early. During exercise, the flow of electrons down the respiratory chain quickens, as does oxygen consumption. The overall effect is greater oxidation of complex I, and lower leakage. — A fall in the reduction state of complex I explains other apparent anomalies, such as the long lifespan of mice with high resting metabolic rates. Brand, working with John Speakman and colleagues at the University of Aberdeen, showed that these mice had more uncoupling proteins in their mitochondria, enabling electron flow to be uncoupled from ATP production, dissipating energy as heat. Uncoupling meant they consumed more oxygen at rest, yet they lived longer than other mice.2 — Uncoupling may be important in people, too. Mitochondrial DNA haplotypes vary geographically, with some types predominant in tropical regions, others in colder climes. The pattern might reflect differing degrees of uncoupling, restricting internal heat generation in hot climates, and vice versa. A consequence might be a higher rate of ROS leak in tropical peoples, and a correspondingly higher susceptibility to degenerative conditions such as heart disease. — Intervention might be possible. Vladimir Skulachev at Moscow State University points to recent work showing that the reduction state of complex I depends strongly on the NAD+ and NADH levels. “Perhaps we could lower ROS leakage, and correspondingly apoptosis, by maintaining a tighter control over the NADH pool.”
The mitochondrial electron transport chain works less well with aging, and this is associated with increased oxidative damage, in all probability due to increased electron leakage. Further, increasing the efficiency of the electron transport chain can extend lifespansof mice.
Several publications discuss this point including the 2003 publication Mitochondrial electron transport chain complexes in aging rat brain and lymphocytes, the 2007 publication The mitochondrial energy transduction system and the aging process and the 2010 publication Mitochondrial electron transport chain functions in long-lived Ames dwarf mice. Also relevant is the recent 2012 publication RhTFAM treatment stimulates mitochondrial oxidative metabolism and improves memory in aged mice. Free radicals associated with increasing age-related increases in electron leakage seem to be the culprits associated with age-related decline in neural functionality. “Free radical-mediated oxidations are determining factors of mitochondrial dysfunction and turnover, cell apoptosis, tissue function, and lifespan. Inner membrane enzyme activities, such as those of complexes I and IV and mitochondrial nitric oxide synthase, decrease upon aging and afford aging markers. The activities of these three enzymes in mice brain are linearly correlated with neurological performance, as determined by the tightrope and the T-maze tests(ref).”
Since mtDNA spin off so much ROS and are damage-prone and have limited self-repair capability, it seems logical to surmise that what is going on with aging is increasing amounts of mutationally damaged mtDNA and that this is correlated with greatly increased susceptibility to age-related neurological and other diseases. This is actually a quite established view of aging, articulated the years ago in the publication Oxidative Stress, Mitochondrial DNA Mutation, and Impairment of Antioxidant Enzymes in Aging1. “Mitochondria do not only produce less ATP, but they also increase the production of reactive oxygen species (ROS) as by-products of aerobic metabolism in the aging tissues of the human and animals. It is now generally accepted that aging-associated respiratory function decline can result in enhanced production of ROS in mitochondria. Moreover, the activities of free radical-scavenging enzymes are altered in the aging process. The concurrent age-related changes of these two systems result in the elevation of oxidative stress in aging tissues. Within a certain concentration range, ROS may induce stress response of the cells by altering expression of respiratory genes to uphold the energy metabolism to rescue the cell. However, beyond the threshold, ROS may cause a wide spectrum of oxidative damage to various cellular components to result in cell death or elicit apoptosis by induction of mitochondrial membrane permeability transition and release of apoptogenic factors such as cytochrome c. Moreover, oxidative damage and large-scale deletion and duplication of mitochondrial DNA (mtDNA) have been found to increase with age in various tissues of the human. Mitochondria act like a biosensor of oxidative stress and they enable cell to undergo changes in aging and age-related diseases. On the other hand, it has recently been demonstrated that impairment in mitochondrial respiration and oxidative phosphorylation elicits an increase in oxidative stress and causes a host of mtDNA rearrangements and deletions. Here, we review work done in the past few years to support our view that oxidative stress and oxidative damage are a result of concurrent accumulation of mtDNA mutations and defective antioxidant enzymes in human aging.”
Earlier publications relating lifespans of rodents to mitochondrial coupling and electron leakage include: Extension of murine life span by overexpression of catalase targeted to mitochondria (2005) and Uncoupled and surviving: Individual mice with high metabolism have greater mitochondrial uncoupling and live longer (2004).
Mitochondrial mutations seem highly associated with exceptional longevity across a number of species including our own.
Certain mitochondrial mutation polymorphisms appear to be associated with exceptional longevity in human subpopulations. And mitochondrial antioxidant interventions extend the lives of mice and other lower species. Among the publications relating mitochondrial mutation polymorphisms to longevity and disease processes are:
Association of mitochondrial DNA haplogroups with exceptional longevity in a Chinese population.
Mitochondrial DNA polymorphisms associated with longevity in a Finnish population.
Mitochondrial DNA haplogroup D4a is a marker for extreme longevity in Japan.
Association of mtDNA haplogroup F with healthy longevity in the female Chuang population, China.
Mitochondrial DNA haplogroups associated with age-related macular degeneration.
Association of the mitochondrial DNA haplogroup J with longevity is population specific.
Mitochondrial DNA haplogroups in Spanish patients with hypertrophic cardiomyopathy.
Are mitochondrial haplogroups associated with extreme longevity? A study on a Spanish cohort.
Mitochondrial DNA haplogroups: role in the prevalence and severity of knee osteoarthritis.
Some variations in the mitochondrial genome are correlated with disease susceptibilities and aging.
The 2006 publication The role of mitochondria in ageing and carcinogenesis reported: “Mitochondria can perform multiple cellular functions including energy production, cell proliferation and apoptosis. These organelles contain their own genetic material, mitochondrial DNA (mtDNA), which is maternally inherited. Although much smaller than the nuclear genome, mtDNA is equally important, as it has been hypothesized to play a crucial role in ageing and carcinogenesis. This is partly due to the fact that mitochondria represent the major site for the generation of cellular oxidative stress and play a key role in mediating programmed cell death (apoptosis). Damage to mtDNA is therefore an important contributor to human ageing, cancer and neurodegenerative diseases. The most relevant footprints of mtDNA damage are point mutations of single bases, or deletions of the 16.5-kb mitochondrial genome. This review will focus on the key roles of mitochondrial function and mtDNA in oxidative stress production and as a mediator of apoptosis, and on the use of mtDNA as a biomarker of sun exposure. This will be related to the contribution of mitochondria and mtDNA in the ageing process and cancer, with a specific focus on human skin. In conclusion, it is likely that the interplay between nuclear and mitochondrial genes may hold the final understanding of the mitochondrial role in these disease processes.”
The 2011 publication The role of the mitochondrial genome in ageing and carcinogenesis relates: “Mitochondrial DNA mutations and polymorphisms have been the focus of intensive investigations for well over a decade in an attempt to understand how they affect fundamental processes such as cancer and aging. Initial interest in mutations occurring in mitochondrial DNA of cancer cells diminished when most were found to be the same mutations which occurred during the evolution of human mitochondrial haplogroups. However, increasingly correlations are being found between various mitochondrial haplogroups and susceptibility to cancer or diseases in some cases and successful aging in others.”
Other publications discussing the roles of mitochondria in disease processes and aging include:
Mitochondrial DNA content varies with pathological characteristics of breast cancer.
Estrogen receptor mediates a distinct mitochondrial unfolded protein response.
Oxygen tension changes the rate of migration of human skin keratinocytes in an age-related manner.
Potential therapeutic benefits of strategies directed to mitochondria.
Genomic profiling identifies GATA6 as a candidate oncogene amplified in pancreatobiliary cancer.
Mitochondrial mutation diseases provide other models of how dysfunctional mitochondria lead to pathologies.
There are perhaps 50 such diseases, mostly rare ones that show up in infants and children. There is a substantial number of research publications about them, most of which are how about a certain mutation correlates with its pathological manifestations. The diseases of these mitochondrial DNA mutations appear to lead to all kinds of disease manifestations: neurological, immunological, cardiovascular, etc. Examples of these publications are:
Mitochondrial tRNA(Phe) mutation as a cause of end-stage renal disease in childhood, A novel mutation in the mitochondrial tRNA(Ser(AGY)) gene associated with mitochondrial myopathy, encephalopathy, and complex I deficiency.
Infantile presentation of the mtDNA A3243G tRNA(Leu (UUR)) mutation.
Studies of such rare diseases may provide clues as to what is going on in major neurological diseases such as ALS, Huntington’s disease (HD), Multiple Scleroris (MS), Parkinson’s Disease and Alzheimer’s Disease – and even autism. The root cause is thought to be mitochondrial dysfunction resulting from mtDNA damage and leading to excess levels of reactive oxygen and nitrogen species.
This point is discussed in the publication Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. “There is significant evidence that the pathogenesis of several neurodegenerative diseases, including Parkinson’s disease, Alzheimer’s disease, Friedreich’s ataxia (FRDA), multiple sclerosis and amyotrophic lateral sclerosis, may involve the generation of reactive oxygen species (ROS) and/or reactive nitrogen species (RNS) associated with mitochondrial dysfunction. The mitochondrial genome may play an essential role in the pathogenesis of these diseases, and evidence for mitochondria being a site of damage in neurodegenerative disorders is based in part on observed decreases in the respiratory chain complex activities in Parkinson’s, Alzheimer’s, and Huntington’s disease. Such defects in respiratory complex activities, possibly associated with oxidant/antioxidant imbalance, are thought to underlie defects in energy metabolism and induce cellular degeneration. The precise sequence of events in FRDA pathogenesis is uncertain. The impaired intramitochondrial metabolism with increased free iron levels and a defective mitochondrial respiratory chain, associated with increased free radical generation and oxidative damage, may be considered possible mechanisms that compromise cell viability. Recent evidence suggests that frataxin might detoxify ROS via activation of glutathione peroxidase and elevation of thiols, and in addition, that decreased expression of frataxin protein is associated with FRDA. Many approaches have been undertaken to understand FRDA, but the heterogeneity of the etiologic factors makes it difficult to define the clinically most important factor determining the onset and progression of the disease. However, increasing evidence indicates that factors such as oxidative stress and disturbed protein metabolism and their interaction in a vicious cycle are central to FRDA pathogenesis. Brains of FRDA patients undergo many changes, such as disruption of protein synthesis and degradation, classically associated with the heat shock response, which is one form of stress response. Heat shock proteins are proteins serving as molecular chaperones involved in the protection of cells from various forms of stress. In the central nervous system, heat shock protein (HSP) synthesis is induced not only after hyperthermia, but also following alterations in the intracellular redox environment. The major neurodegenerative diseases, Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), Huntington’s disease (HD) and FRDA are all associated with the presence of abnormal proteins. Among the various HSPs, HSP32, also known as heme oxygenase I (HO-1), has received considerable attention, as it has been recently demonstrated that HO-1 induction, by generating the vasoactive molecule carbon monoxide and the potent antioxidant bilirubin, could represent a protective system potentially active against brain oxidative injury. Given the broad cytoprotective properties of the heat shock response there is now strong interest in discovering and developing pharmacological agents capable of inducing the heat shock response. This may open up new perspectives in medicine, as molecules inducing this defense mechanism appear to be possible candidates for novel cytoprotective strategies. In particular, manipulation of endogenous cellular defense mechanisms, such as the heat shock response, through nutritional antioxidants, pharmacological compounds or gene transduction, may represent an innovative approach to therapeutic intervention in diseases causing tissue damage, such as neurodegeneration.” Italics emphasis on the presence of abnormal proteins is our own since it a theme we will pick up later in this discussion.
“Mitochondrial disease can look like any number of better known diseases, including: Autism, Parkinson’s disease, Alzheimer’s disease, Lou Gehrig’s disease, muscular dystrophy and chronic fatigue, among others. Adults and children with it can have features similar to other disorders like: Epilepsy, Myopathy, Developmental Delay, learning disabilities and, Fibromyalgia — Research shows that mitochondrial dysfunction is often a central element of these more commonly recognized diseases. Studies and reports indicate the “orange” ones are more influenced. A cure for mitochondrial disease could impact cures for Autism, Parkinson’s, Alzheimer’s and Muscular Dystrophy(ref).”
With mitochondrial mutation diseases, there is an electron leak rate as high as 40% producing much larger volumes of free radicals outside of the hormetic range.
Again, it seems that Mitochondrial electron leakage is a smoking gun for ROS production that is highly correlated with disease processes and longevity. In this series of blog posts, we explore the nature of some of those correlations. And we discuss practical intervention strategies.
Certain autoimmune and neurological diseases are also characterized by deficiency in electron chain operation, more electron leakage and excessive ROS production.
Here the literature suggesting the problem goes back a bit. The 2006 publication Mitochondria, apoptosis and autoimmunity reports: “– Several mitochondrial proteins have been implicated as regulators of apoptosis in the immune system that are required for prevention of autoimmunity. Recent discoveries have shed light on mitochondrial functions as they relate to cell death, including caspase-dependent and -independent apoptosis, mitochondrial death substrates and events that disable mitochondrial functions during apoptosis. These discoveries, taken with reports that the specific manner by which a cell dies greatly impacts on the nature of subsequent immune responses, highlight an exciting era of research on mitochondrial function and its role in apoptosis and the effects on immune responses.”
The just-published report Sjøgren’s syndrome-associated oxidative stress and mitochondrial dysfunction: Prospects for chemoprevention trials relates: “An involvement of oxidative stress (OS) was found in recent studies of Sjøgren’s syndrome (SS) that reported significant changes in protein oxidation, myeloperoxidase activity, TNF-α, nitrotyrosine and GSH levels in plasma from SS patients. Excess levels of OS markers, as oxidative DNA damage and propanoyl-lysine were reported in saliva from SS patients. Previous reports concurred with a role of OS in SS pathogenesis, by showing a decreased expression of antioxidant activities in conjunctival epithelial cells of SS patients and in parotid gland tissue samples from SS patients. A link between OS and mitochondrial dysfunction (MDF) is recognized both on the grounds of the established role of mitochondria in reactive oxygen species (ROS) formation and by the occurrence of MDF in a set of OS-related disorders. Early studies detected mitochondrial alterations in cells from SS patients, related to the action of antimitochondrial autoantibodies, and affecting specific mitochondrial activities. Thus, a link between MDF and OS may be postulated in SS, prompting studies aimed at elucidating SS pathogenesis and in the prospect of chemoprevention trials in SS clinical management.”
It appears that in certain disease processes, induced oxidative stress precipitates a long-lasting chain of stress response events in mitochondria
The just-published (November 2012 document Self-evolving oxidative stress with identifiable pre- and postmitochondrial phases in PC12 cells reports: “During the neurodegenerative process in several brain diseases, oxidative stress is known to play important roles in disease severity and evolution. Although early events of stress, such as increased lipid peroxidation and decreased superoxide dismutase, are known to characterize early onsets of these diseases, little is known about the events that participate in maintaining the chronic evolving phase influencing the disease progression in neurons. Here, we used differentiated PC12 cells to identify premitochondrial and postmitochondrial events occurring during the oxidative stress cascade leading to apoptosis. Our data indicate that an acute and strong oxidative impulse (500 μM H(2) O(2) , 30 min) can induce, in this model, a 24-hr self-evolving stress, which advances from a premitochondrial phase characterized by lysosomes and cathepsin B and D translocations to cytosol and early mitochondrial membrane hyperpolarization. This phase lasts for about 5 hr and is followed by a postmitochondrial phase distinguished by mitochondrial membrane depolarization, reactive oxygen species increase, caspase-9 and caspase-3 activations, and apoptosis. Inhibition of cathepsins B and D suggests that cells can be protected at the premitochondrial phase of stress evolution and that new cathepsins regulators, such as glycosaminoglycans mimetics, can be considered as new therapeutic prototypes for neurodegeneration. Insofar as early oxidative stress markers have been related to the early onset of neurodegeneration, strategies protecting cells at the premitochondrial phase of oxidative stress may have important therapeutic applications.
Since mitochondria are short-lived and mtDNA is so subject to mutational damage, mitochondrial biogenesis is an important consideration.
Since I have discussed mitochondrial biogenesis before I will treat it only in a summary fashion here. From Wikipedia: “Mitochondrial biogenesis is the process by which new mitochondria are formed in the cell. Mitochondrial biogenesis is activated by numerous different signals during times of cellular stress or in response to environmental stimuli. — It is reckoned that higher mitochodrial copy number (or higher mitochondrial mass) is protective for the cell. — Mitochondria are produced from the transcription and translation of genes both in the nuclear genome and in the mitochondrial genome. The majority of mitochondrial protein comes from the nuclear genome, while the mitochondrial genome encodes most parts of the electron transport chain along with mitochondrial rRNA and tRNA. A major adaptation to mitochondrial biogenesis results in more mitochondrial tissues which increases metabolic enzymes for glycolysis, oxidative phosphorylation and ultimately a greater mitochondrial metabolic capacity.[Peter D. Wagner Summer 2011 Lecture]– The master regulators of mitochondrial biogenesis appear to be the peroxisome proliferator-activated receptor gamma (PGC) family of transcriptional coactivators, including PGC-1α, PGC-1β, and the PGC-related coactivator, PRC. PGC-1α, in particular, is thought to be a master regulator. It is known to co-activate nuclear respiratory factor 2 (NRF2/GABPA), and together with NRF-2 coactivates nuclear respiratory factor 1 (NRF1). The NRFs, in turn, activate the mitochondrial transcription factor A (tfam), which is directly responsible for transcribing nuclear-encoded mitochondrial proteins. This includes both structural mitochondrial proteins as well as those involved in mtDNA transcription, translation, and repair.”
I have discussed PGC-1α and Nrf2 in a number of blog entries related to mitochondrial biogenesis. In a June 2010 blog entry AMPK and longevity, I discussed how exercise activates the AMPK pathway and the role of PGC-1alpha (peroxisome-proliferator-activated receptor gamma co-activator-1alpha) as a co-transcriptional regulation factor that induces mitochondrial biogenesis by activating transcription factors. The August 2010 blog entry PGC-1alpha and exercise provides a further and more general introduction to PGC1alpha. I said “You can probably expect to hear a lot about PGC-1alpha as time goes on because this remarkable substance is turning out to have a lot to do with health and longevity. It appears to be the mediator of the health benefits produced by exercise. This blog post is about PGC-1alpha, about its relationship to exercise, and about efforts to stimulate it with various substances, in essence seeing if it is possible to provide “exercise in a pill.” And in the blog entry PQQ – activator of PGC-1alpha, SIRT3 and mitochondrial biogenesis I discuss how Pyrroloquinoline quinone (PQQ), a redox cofactor available as a dietary supplement, appears to have at least three central biological effects with powerful downstream health and longevity consequences: it stimulates the generation of PGC1-alpha, results in expression of SIRT3, and induces mitochondrial biogenesis. If there is such a thing as exercise in a pill, it is probably PQQ.

Image source: James P Watson. Since the image source raises certain critical questions, Jim is not sure it is helpful. The questions Jim identified are:
- What part is important?
- What changes with aging?
- Can we affect mitochondrial ROS and health?
- What can you do about it?



Image source: James P Watson. Jim comments: “Mitochondrial Biogenesis and Reactive Oxygen SpeciesShown is mitochondrial biogenesis during calorie restriction versus ad libitum feeding in mice and its proposed effects on reactive oxygen species (ROS). In the ad libitum case, the number of electron transport chains is low, and if the rate of entry of electrons (red e−) exceeds the slowest step of flow through the chain, stalling of electrons at mitochondrial complexes I and III (blue e−) and production of ROS will be favored. During calorie restriction, mitochondrial biogenesis increases the number of electron transport chains, thereby reducing the rate of electron entry per electron transport chain. Calorie restriction may also increase the fraction of electrons that bypass complex I by entering the electron transport chain via the electron transfer flavoprotein dehydrogenase (ETF). These effects may reduce the production of ROS during calorie restriction and hence mitigate cellular damage, aging, and disease. Reference: Leonard Guarante, Mitochondria- A Nexus for Aging, Calorie Restriction, and Sirtuins?, Cell, Vol 132(2), pp171-176, January 25, 2008”
Mitohormesis is another important concept relating to oxidative stress, health of mitochondria and probably organism longevity.
I have devoted a recent blog entry to this topic Mitohormesis, so will devote only a few words to it here. The central concept is that energy metabolism in mitochondria produce reactive oxygen species ROS, due to electron leakage and other factors. At a low level these ROS perform essential cell signaling functions, including triggering of NRF2 signaling which activates the body’s endogenous antioxidant defense system. Numerous health-producing effects result as well as demonstrated longevity-related impacts in lower species. This signaling is a phenomenon evolutionarily conserved across a variety of species. Consuming antioxidant supplements can sometimes interfere with such signaling and have negative biological impacts. However, if the oxidative stress is to great, such as might be the case due to excessive electron leakage in the mitochondrial electron transport chain due to age-related mitochondrial gene mutations, in that case the benefits of the signaling might be outweighed by the destructive effects of the ROS. In such a case exogenous antioxidant administration might be a good thing.
A key to understanding what is going on in hormesis is understanding how the hormetic dose-response curve works, and this can be formulated in terms of NRF2 expression.
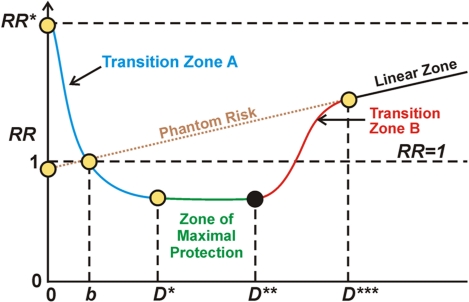
Graphic depiction of the stochastic hormetic relative risk model as given in Scott et al. (2009).
Understanding the typical dose-response curve associated with hormesis is critical for interpreting seemingly contradictory research. In my interpretation, the horizontal axis depicts level of stress, say as driven by ROS load. The vertical axis represents relative risk, level of probable pathological organismic response where normal level is 1. To the left of the first axis crossing in the diagram (point D) the Keap1-Nrf2 pathway is progressively kicking in but not sufficiently so as to overcome the direct negative effects of ROS stress. So, in Transition zone A there is under-expression of the ARE genes and a negative health condition Between stress levels b and D*** there is hormetic protection compared to what would be expected given a linear model of negative response to stress and better-than-expected health, due at least in part to activation of Nrf2 and the ARE genes. The zone of maximum protection is between D* and D**. Starting at D** to D*** the stress load begins to overwhelm the defensive activities of the ARE genes and the protection becomes less and less until at point D*** the hormetic response associated with ARE gene activation becomes negligible. In the case of radiation damage at least, beyond point D*** the damage according to conventional wisdom is in linear proportion to the stressor, the amount of radiation. Phantom risk is theoretical risk for low stress levels that would apply if the linear model were extrapolated for low stress dosages.
For further discussion of this curve and aspects of Mitohormesis, please see the Mitohormesis blog entry.
Moving forward
This blog entry summarizes some basic information about mitochondria, some found in textbooks and some not. By now it should be clear that mitochondria play central roles related to biology in general, health and longevity. This blog entry is only the appetizer with the main course to follow. As I now see it, the next blog entry in this series, Mitochondria Part 2: mitochondrial pathways, diseases and aging will dig deeper and examine underlying pathway mechanisms for observed disease and aging effects. We will touch upon the sources and natures of mitochondrial stress. We go some into mitochondrial-nuclear signaling and the mitochondrial UPR (unfolded protein response), the roles of mitochondria in UPR in the endoplasmic reticulum, and into signaling transport mechanisms such as microtubules and motor proteins. And we will speculate on a possible underlying mitochondrial cause common to aging and age-related neurological, cardiac and other diseases. As I see it now, a Part 3 blog entry will focus on practical interventions. It will review what is known about the impact of administration of key mitochondrial antioxidants, alpha-lipoic acid, acetyl-l-carnitine, melatonin and buckyballs. And we will speculate on how further research needs to be directed to answer critical outstanding questions.

{kind=link}
Hi
This is interesting — but some of the figures showing mitochondrial systems are too blurry to read on my computer. I look forward to seeing the future posts on this subject — hopefully with readable figures.
Jim Rose
Hi jhrose
Thanks for the feedback. I had a problem getting adequate resolution on some of the images Jim Watson sent me in a PowerPoint presentation he was putting together. Getting them online was an awkward multistep process. I will see if I can upgrade the images and take measures to assure that future images are fully readable. We are working on the Part2 posting in this blog series, and I think it will include some exciting stuff.
Vince
Pingback: Multifactorial hormesis – the theory and practice of maintaining health and longevity | AGING SCIENCES – Anti-Aging Firewalls
Pingback: Futureseek Daily Link Review; 28 March 2013 | Futureseek Link Digest
Pingback: Autophagy – the housekeeper in every cell that fights aging | AGING SCIENCES – Anti-Aging Firewalls
Pingback: Unlocking Longevity - AGINGSCIENCES™ - Anti-Aging Firewalls™