
By James P Watson with assistance by Vince Giuliano
This blog entry looks at the genetic and epigenetic factors in cancer. The news here is basically good – epigenetic transformations seem in many cases to be upstream of genetic mutations and there seem to be interventions that can reverse such transformations in the long periods of pre-cancerous development. Certain cancers may well be untreatable or require brutal drastic treatments, but they might be preventable in the first place. The blog entry also looks at the roles of P53 and of exosome communications in the development, prevention and proliferation of cancers. Here, the news is mixed – partially positive and partially negative because cancer cells can use exosome communications just like our own cells do. The blog relates an up-to-date interweaving of results from several newer fields of research: exosome molecular communications, cancer genetics and epigenetics, cancer gene silencing and activation pathways, and cell senescence theory. It even talks about secreted frizzled proteins!
Genetic mutations and epigenetic shifts in cancer
The traditional view is that cancers are due to genetic mutations – ones that occur as a result of behaviors like cigarette smoking, or ones that occur randomly such as errors in DNA repair and anuploidy. Once those mutations are there, nothing can be done about them. End of story.
But that is actually only part of the story. Epigenetic mutations tend to far outnumber the genetic ones in any cancer and tend to be upstream of the genetic mutations and show up prior to them before full-scale cancers develop. And we can probably do something about them leading to cancer prevention, for example via administration of phytochemicals. In more detail, let’s start with the conventional explanations for DNA mutations:
- Spontanous CpG mutations – Spontaneous deamination of cytosine bases in the DNA-binding domain of genes, inducing a C to A substitution in the DNA base. Both cytosine and methylated cytosine undergo this spontaneous deamination, but methylated cytosine is less stable and has a 5-fold higher rate of spontaneous deamination, compared to T,G, or A bases. As a result, CpG dinucleotides mutate at a rate that is 10 times higher than other nucleotides. Consequently, the frequency of CpG dinucleotides in our genome is lower than expected and is thought to be due to this spontaneous deamination of the germ line during evolution. Only half of our genes have CpG islands in their promoter regions. In the religious interpretation, “Sin destroys your epigenetic marks”. In the evolutionary interpretation, “evolution changes your epigenetic marks to induce evolution” (I buy the evolution explanation over the Satan explanation!) This spontaneous deamination is driven by high baseline ROS and RNS most commonly seen in the setting of age-induced chronic inflammation. Chronic inflammation is the hallmark of aging. Thus “C-to-A substitutions are due to numerous-birthday toxicity.”
One of the key ideas here is that species need mechanism for slightly shaking up the genomes of succeeding generations to allow genetic evolution to occur in the face of changing circumstances. One of several such mechanisms could be the spontaneous GpG mutations.
References: 1. Bird A.P. CpG-rich islands and the function of DNA methylation. Nature1986;321:209-213.
2. Jones PA, Rideout WM, Shen JC, Spruck CH, Tsai YC. 1992. Methylation, mutation and cancer. Bioessays 14: 33–36.
- Cigarette smoke – Smoke enters our lungs without being detoxified by our liver. Bypassing the liver is the main reason why inhaling smoke is so bad. Polycyclic aromatic hydrocarbon (PAH) induced epoxidation of DNA, which preferentially induces a G to T change in the DNA-binding domains of genes. With the p53 gene, this occurs at codons 157, 158, and 273.
Thus “smokers like to put epoxy glue on their guanines”. (or something like that….read the reference below). This explains lung cancers and other smoking-related diseases. Thus “G-to-T changes are due to Cigarettes”.
Reference: 1. Pfeifer GP, Hainaut P. 2003. On the origin of G –>T trans-versions in lung cancer. Mutat Res 526: 39–43.
2. Pfeifer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht SS, Hainaut P. 2002. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking- associated cancers. Oncogene 21: 7435–7451.
Having the reputation of “guardian of our genome,” P53 has long been known to be one of our major bulwarks against cancer, so mutating it and taking it out of action is not a good thing. One of the actions P53 is well known for in cancer cells is induction of apoptosis. P53 has other important actions with respect to cancer however, and some of these are reviewed here below.
- Diesel smoke – Diesel smoke also enters our lungs without being detoxified by our liver. The carcinogen in diesel smoke, 3-NBA, induces nitroreduction of DNA, which preferentially induces a G to T change in the DNA-binding domains of genes. Thus, “G-to-T changes are due to driving behind a diesel truck”.
- Sun – The sun “picks on” DNA where there are two cytosines side-by-side (i.e. a CC). Specifically, UVB and UVC light induce a CC to TT change in the DNA-binding domains of genes. In the p53 gene, this occurs at codons 248 and 278. In other words, “the sun likes to pick on CC twins“. If your gene has a “CC twin”, then don’t become an Egyptian (they worshipped the sun!)
Reference: Giglia-Mari G, Sarasin A. 2003. TP53 mutations in human skin cancers. Hum Mutat 21: 217–228.
Of course, there is a long additional list of stressors known to induce cancer gene mutations like toxic metals and chemicals. Most act via super-hormetic doses of reactive oxygen species, but these above are representative.
5. Epigenetic silencing – One way of silencing a cancer-protective gene like P53 is by mutating it. But a gene can also be effectively silenced via epigenetic mechanisms including DNA Methylation, Histone deacetylation, post-translational attachments of siRNAs and silencing of important upstream pathway genes. The end result is the same – the protein corresponding to the gene does not get made.
DNA Methylation is a broad and important topic discussed before in this blog(in about 80 posts starting with this one in 2009) and extensively elsewhere. E.g. (ref)(ref). As a general comment, with aging there tends to be hypermethylation of genes with GPC islands leading to decline in their over-expression, and global hypomethylation of other genes leading to unwanted over-expression of them. Due to imperfect DNA methylation maintenance, hypermethylation of CpG islands occurs due both DMMT1 and DNMT3b. Once they are hypermethylated, methyl binding proteins (MBDs) sit on the methylated CpG islands and prevents their demethylation. MBDs are more or less “permanent residents” on the hypermethylated CpG islands, where as the other factors here are “shuffling around” the CpG islands. The “shufflers” include HDACs, which deacetylate H3K9 histone marks. Following H3K9 demethylation, DNMTs also recruit HMTs which methylate H3K9 histone marks. The opposite occurs for H3K4 – this one is demethylated with gene silencing. Polycomb proteins are also “shuffling around” and are involved with CpG island gene silencing.
Reference: Epigenetic gene silencing in cancer: the DNA hypermethylome
6. Also possibly implicated in cancers is the loss of epigenetic silencing of coding genes and non-coding DNA that promote cancer, such as due to micro satellite regions and Alu repetitive elements, resulting in the amplification of these genes and non-coding DNA expression.
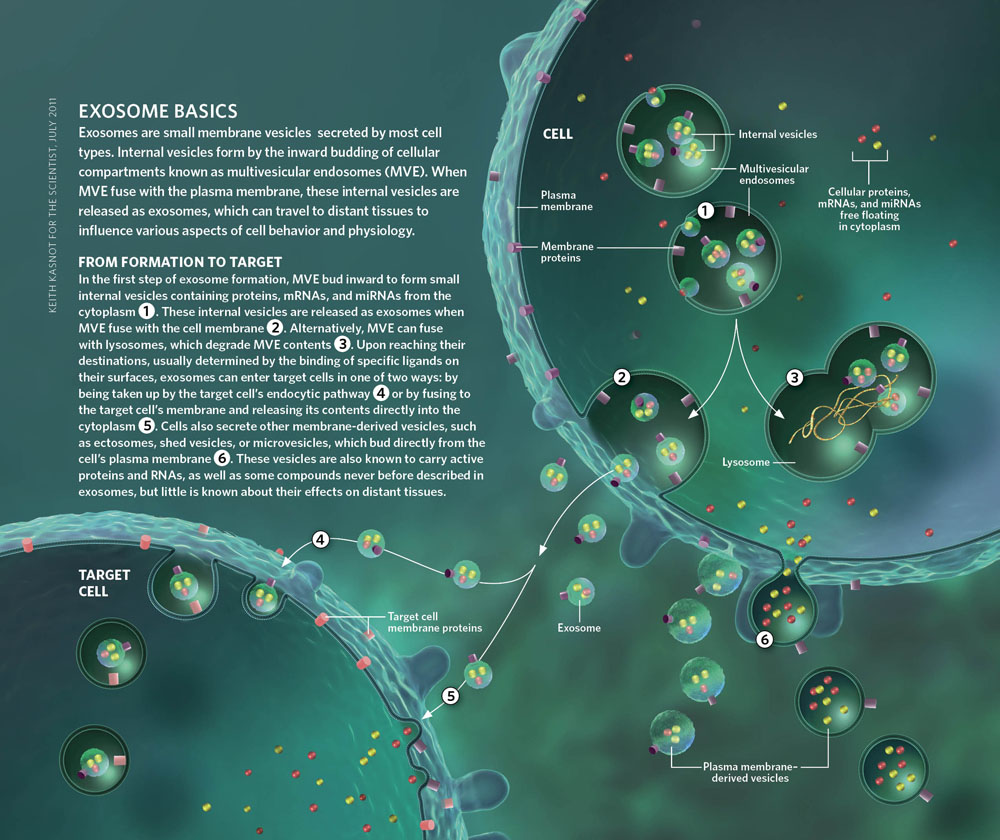
A few basics about exosomes
Exosomes(ref)(ref)(ref)(ref) are tiny vesicles generated within cells that can contain multiple substances (e.g. mRNAs, ncRNAs, proteins) and that can be critical for multiple important functions including intra and inter-cellular signaling, and cell “trash removal.” They are found in eukaryotes and archaea and are secreted by many cell types in a wide variety of organisms. They are secreted by many cell types including stem cells in a wide variety of organisms and appear to be a very early evolutionary adaption for signaling needs in multi-cell organisms. One of their important roles is serving as receptor-addressable information packets capable of protecting signaling molecules and readily transferring them across membrane barriers. Exosomes are thought to be produced via the formation of multivesicular bodies and secreted through the plasma membrane via exocytosis. They are taken into target cells via endocytotic pathways. They are very tiny (30-200nm) and sufficiently small that their migration can be activated by brownian motion at normal physiologic temperatures. They perform a number of signaling functions critical to numerous essential cell functions. They are capable of transporting signaling molecules that direct and control cellular adaptions to stresses and gene regulation. Their production appears to be induced by stresses.
The probable role of exosomes in quorum sensing and interspecies communications was touched on in the blog entry Quorum Sensing Part 2 – Intra and inter-species molecular communications, Under settings of severe stress such as due to an infection, they can be present in very large numbers – three orders of magnitude more than numbers of red blood cells, for example. Exosomes readily cross the blood-brain barrier – two order of magnitude more readily than most drugs and proteins for example. The research literature related to exosomes goes back to the 1980s, though the number of publications has risen steeply in recent years. There are now 1335 publications related to exosomes according to pubmed.org. As discussed below, they are used by cancer as well as healthy cells and therefore also have a dark side. Finally, there is much more that can be said about exosomes beyond the cancer-related aspects discussed here.
1. No Stress –> No Exosomes
Cellular stress triggers p53 activation which induces the secretion of free proteins and exosome-bound proteins. The free proteins secreted and the proteins secreted inside exosomes have an inhibitory role in cancer
The triggers for p53 activation
It is now quite well established that cellular stress induces the transcription factor p53, in response to X-rays, gamma rays, UV light, heat, hypoxia, chemotherapy, and activated oncogenes. In addition to its role as a tumor suppressor via its role in apoptosis, cell cycle arrest and senescence, p53 also regulates the synthesis and secretion of proteins and exosomes from the stressed cell. The role of p53 as the “master exosome secretor” was not known until 2006.
Reference: Yu et al, Cancer Research, 2006, The Regulation of Exosome Secretion: a Novel Function of the p53 Protein
Proteins secreted in response to p53 activation (aka classical secretion) – IGF-BP3, PAI-1, and thrombospondin
The secreted proteins are expressed and secreted in response to p53 activation via the classical secretion mechanism. Proteins secreted free (not in exosomes) can be identified by the presence of a “signal sequence” at the NH2 terminus of the protein. The “signal sequence” directs the protein through the endoplasmic reticulum, then the Golgi apparatus, then to the plasma membrane via vesicles. This “signal sequence” can be used to identify which proteins that increase with p53 activation are secreted free, without being contained inside exosomes. The p53 induced proteins secreted this way include IGF-BP. PAI-1, and thrombospondin. There are specific purposes for these free, secreted proteins. IGF-BP3 down-regulates the insulin/IGF pathway. PAI-1 inhibits proteases that alter the ECM, and thrombospondin blocks angiogenesis. These free proteins secreted by the stressed cell act to produce more coordinated response to stress…… a sort of quorum sensing function like that seen in bacteria that are stressed(ref).
Exosome secreted in response to p53 activation (aka non-classical secretion)
In unstressed cancer cells, exosome secretion occurs but is in such low levels that they are barely detected. The same phenomena appears to be true in stem cells such as embryonic or mesenchymal stem cells (no stress => no exosomes). However with any of the cellular stressors described above (4 G of XRT, gamma rays, UV light, hypoxia, chemotherapy, or heat), exosome secretion occurs. This is a p53 mediated phenomena and occurs as a result of the activation of the TSAP6 gene. Several proteins without a “signal sequence” at the NH2 terminus of the protein are secreted in these exosomes. (This is how you can tell if a protein is secreted “free” or if it is secreted inside an exosome).
Their synthesis may or may not increase inside the cell, but their secretion always increases with p53 induction. The p53 induced proteins secreted in exosomes include Maspin, CyPA, PRDX-1, PGK1, Enol translation, EF-1a, and Hsp90beta proteins. Maspin has an inhibitory effect on angiogenesis, tumor invasion, and metatastisis.
Reference: The Regulation of Exosome Secretion: a Novel Function of the P53 Protein
Conclusion: Cellular stress is clearly the cause of exosome secretion. “No stress => No exosomes!”
p53 induces both “free protein secretion” via the classical pathway and “exosome bound protein secretion” via the non-classical pathway. Not all proteins concentrated in exosomes are found in increased concentrations inside the cell.
2. No p53 => No Exosomes
25-50% of cancers have a mutated p53 gene.
p53 mutations – mostly missense mutations at CpG residues between codons 125 and 300 due to free radical induced deamination of methylated cytosine, seen in conditions of chronic inflammation
Many human cancers have p53 mutations. 73% of these mutations are “missense mutations”, which means they change the amino acid sequence of p53. The most common mutation is a G:C to an A:T at a CpG site, which is usually associated with epigenetic methylation. This occurs because methylated cytosine is 5 times more likely to undergo spontaneous deamination into thymine than an unmethylated cytosine base. This “spontaneous deamination” is very predictable – it occurs due to oxygen and nitrogen free radicals seen in high concentrations with chronic inflammation. Thus, chronic inflammation is the greatest “trigger” of mutations and these mutations show up at methylated cytosine bases in genes. The mutations are not randomly distributed throughout the 380 codons in the TP53 gene. Instead, most of them are clustered in the DNA binding domain (DBD), which is between codons 125 and 300. Of the 22 CpG sites in this DNA-binding domain, 3 “hotspot codons” represent 60% of the CpG mutations and another five residues account for 26% of these mutations. The lack of mutations at other sites does not mean that they do not occur……it just means that mutations at those other sites do not generate a dysfunctional protein.
In cancer cells without a functional p53, no exosome secretion occurs. Thus it is clear that exosome secretion is due to p53. This fact is extremely important, since tests are being developed now for the detection of cancer using exosomes. Since somatic mutations in the p53 gene is the MOST COMMON MUTATION SEEN IN CANCER, using exosomes as biomarkers for cancer will NOT be accurate in the cancers with mutated p53.
Mutations in p53 are more frequent in two scenarios:
1. More aggressive tumors – thus, exosomes will NOT be a good biomarker of tumor aggressiveness
2. More advanced stage – thus, metastatic tumors will be less likely to secrete exosomes.
These facts have tempered the enthusiasm for exosome biomarker research. Here are the rates of p53 mutations in various cancers (which will confer an equivalent percentage of “false negatives” with exosome biomarkers for these tumors):
Reference: P53 Mutations in Human Cancers: Origins, Consequences and Clinical Use
Accelerated p53 degradation in cancer
Even in tumors with no p53 mutations, p53 is not always expressed normally or may be degraded too fast. For instance, in cervical cancers caused by the HPV virus, p53 is functional, but is rapidly degraded by the HPV E6. In sarcomas, the HDM2 gene is over-expressed. This also results in accelerated degradation of p53. Thus the absence of a p53 mutation does not necessarily mean that p53 function is normal.
3. The Hypermethylome – Promoter CpG hypermethylation is a hallmark of all cancers
relevance to exosomes: The p53 gene (TP53) does not have a CpG island, but several genes that make up the p53 pathway do have CpG islands. They include p14ARF, p73, and HIC-1. If one or more of these genes are epigenetically silenced, tumors will not secrete exosomes.
DNA gene mutations vs Epigenetic silencing of non-mutated genes
Cancer is a disease that was once thought to be due primarily to DNA mutations in tumor suppressor genes and oncogenes, as well as chromosomal abnormalities such as aneuploidy. This simplistic concept of cancer is probably erroneous or incomplete, however, as more and more discoveries are made about epigenetic silencing of non-mutated genes as well as RNA interference of gene expression on a post-transcriptional level. The most common methods by which non-mutated genes can be silenced is by methylation of cytosine residues of DNA in specific areas of promoter regions that are called “CpG islands”. This is mediated by DNA methyltransferase 1 (DNMT1) and is most often associated with histone modifications associated with gene silencing, such as histone deacetylation of H3 and H4 tails. This method of gene silencing is thought to be responsible for much of the tumor suppressor gene silencing that occurs in non-mutated DNA.
Oncogenes can also be over-expressed by hypomethylation of DNA and histone modifications that increase transcription of a gene. This method of a “loss of gene silencing” is thought to be responsible for much of the oncogene over-expression seen in non-mutated DNA.
The 3D graph below shows the “hypermethylome” of 20 different genes in 18 different categories of cancers.

Image source: Epigenetic gene silencing in cancer: the DNA hypermethylome
Epigenetic silencing is the “chicken” and DNA mutations are the “egg” – epigenetic alterations appear often before DNA mutations in “pre-cancer”
It now appears that epigenetic silencing is an early event in carcinogenesis and may occur in the pre-malignant phases of cancer (i.e. carcinoma-in-situ stage). For instance, an “epigenetic switch” in gene-imprinting status in renal cells may contribute to the early abnormal expansion of cells in Wilms tumor. Epigenetic silencing of several genes appears in the early stages of colon cancer. The epigenetic silencing of the cell cycle controller, p16, has been seen in pre-invasive breast tumors and lung tumors. In the pre-invasive stages of prostate cancer, the loss of expression of glutathione S-transferase (GSTP1) has been seen. Whereas most DNA mutations are thought to be “stochastic events” (i.e. random), epigenetic silencing may not be “stochastic” and may instead occur simultaneously in several genes due to the over-activation of DNMT1 or HDAC enzymes.
Epigenetic silencing may have a “permissive effect” on cancer as well. For instance, if a tumor suppressor is epigenetically silenced, cancer cells may be able to escape senescence. This is seen in the epigenetic silencing of the gene CDKN2A, which encodes for P16(Ink4a). Another example of this “permissive” effect of epigenetic silencing is seen in the silencing of genes involve with DNA repair. When these genes are silenced, DNA mutations may occur. This is seen in the gene MGMT, which is involved with the repair of the DNA base guanosine. When this enzyme is epigenetically silenced, DNA mutations in guanosine bases cannot be repaired.
All of these phenomena clearly show how epigenetic silencing precedes DNA mutations in most pre-cancerous tumors. It is no surprise then, that the cancer-prevention effects of fruits and vegetables are largely mediated by epigenetic effects, not anti-oxidant effects. For instance, there is clear evidence that plant-derived polyphenols prevent DNA methylation of CpG islands, prevent the deacetylation of H3K9 and H3K14 lysine residues on histones, and inhibit microRNA that promote cancer. This has been shown for many polyphenols such as EGCG, genistein, curcumin, etc. If epigenetic silencing is the “chicken”, then here is a list of the “chicken genes” that are epigenetically silenced.
Reference: Epigenetic gene silencing in cancer: the DNA hypermethylome
Pathways Representative hypermethylated genes
DNA repair hMLH1, MGMT, WRN, BRCA1
Hormone response Estrogen, progesterone, androgen, prolactin and thyroid-stimulating hormone receptors
Vitamin response RARB2, CRBP1,
Ras signaling RASSFIA, NOREIA
Cell cycle p16INK4a, p15INK4b, Rb
P53 network p14ARF, p73, HIC-1
Cell adherence and invasion E-cadherin, H-cadherin, FAT, cadherin, EXT-1, SLIT2, EMP3
Apoptosis TMS1, DAPK1, WIF-1, SFRP1
Wnt signaling APC, DKK-1, IGFBP-3
Tyrosine kinase cascades SOCS-1, SOCS-3, SYK
Transcription factors GATA-4, GATA-5, ID4
Homeobox genes PAX6, HOXA9
Other pathways GSTP1, LKB1/STK11, THBS-14, COX-2, SRBC, RIZ1, TPEF/HPP1, SLC5A8, Lamin A/C
microRNAs miR-127 (targeting BCL6), miR-124a (targeting CDK6)
Conclusions from the list above: As you can see from this list above, the p53 gene is not on the list. This is because the p53 gene has no CpG island. (i.e. it is not epigenetically silenced). On the other hand, 3 genes involved with the p53 network can be epigenetically silenced (P14ARF, p73, and HIC-1). Several other interesting things are present in the list above – the genes involved with cellular senescence can be silenced (p16INK4a, Rb, etc.). This may explain why cellular senescence does not always work to “turn off cancer.”
Most interesting to me is that the gene for E-cadherin can be epigenetically silenced via promoter hypermethylation. This explains why Vitamin D cannot “prevent” or “cure” all cancers, since the E-caherin gene is under the regulation of Vitamin D3. Last of all, this list includes several non-coding genes – the microRNAs for miR-127 and miR-124a, which normally regulate Bcl-6 and CDK6. This shows that both coding and non-coding genes can be silenced by epigenetic mechanisms.
HCT116 Colon cancer – a “case study” in the contributions of both DNA mutations and epigenetic silencing
The HTC116 colon cancer cell line is a good example of the relative contributions of both DNA mutations and epigenetic silencing of genes. In HCT116, one of the two copies of the gene for DNA mismatch repair (MLH1) is mutated. As a result, microsatellite regions in genes expand, which is a common phenotype in colon cancer. The gene p16 is also mutated, which also contributes to the “mismatch repair phenotype” as well as to the disruption of the Cyclin D-RB1 cell cycle control pathway. As a result, cells do not have good control over the cell cycle. The gene for TGFbeta2R is also mutated, which results in the loss of control of a pathway involved with cell differentiation. As a result, the cancer cells do not differentiate, which normally would shut off the cell cycle. In addition, a mutation in the gene that encodes for beta-catenin (CTNNB1) results in over-activation of Wnt signaling, which increases cell proliferation. In addition to the 5 mutations above, 14 epigenetically silenced genes have been found in HCT116 (and this is thought to be an incomplete list). As you can see from the list below, one allele of a gene can be mutated and the other one can be epigenetically silenced. Thus, DNA mutations and DNA hypermethylation of CpG islands can work synergistically in cancer.
“Table 1 | Mutated and hypermethylated genes in colon cancer cells*
| Pathway or Function | Hypermethylated genes | Mutations | Biological effects |
| Wnt signalling | SFRP1, SFRP2, SFRP4and SFRP5 | Activating mutation in CTNNB1 | Pathway activation; stem-cell and progenitorcellexpansion; cell survival |
| Mismatch repair | Wild-type MLH1 allele | Second MLH1 allele | Defects in DNA mismatch-repair |
| Cell-cycle regulation | Wild-type CDKN2A allele | Second CDKN2A allele | Blocks cyclin D–RB1 pathway, which results incell proliferation |
| Epithelial-cell differentiation | GATA4, GATA5, TFF1,TFF2, TFF3 and INHA | TGFBR2 | Loss of normal differentiation |
| p53-mediated DNAdamage response | HIC1 | Loss of apoptosis response to DNA damage | |
| Cell invasion | TIMP3 | Loss of inhibition of matrix metalloproteinaseenzymes, which promotes cell invasion |
*Partial list of genetic mutations and heritable gene-silencing events that were identified in a single culture line (HCT116) of human colon cancer cells. CDKN2A, the gene that encodes p16; CTNNB1, the gene that encodes -catenin; GATA, genes that encode GATA-binding transcription factors; HIC1, hypermethylated in cancer 1; INHA, inhibin-; MLH1, a DNA mismatch-repair protein; RB1, retinoblastoma 1; SFRP, secreted frizzled protein; TFF, trefoil factor; TGFBR2, gene that encodes the transforminggrowth-factor-receptor 2; TIMP3, tissue inhibitor of metalloproteinase 3.” Reference: Epigenetic gene silencing in cancer – a mechanism for early oncogenic pathway addiction?
“ Chromatin alterations have been associated with all stages of tumour formation and progression. The best characterized are epigenetically mediated transcriptional silencing events that are associated with increases in DNA methylation — particularly at promoter regions of genes that regulate important cell functions. Recent evidence indicates that epigenetic changes might ‘addict’ cancer cells to altered signal-transduction pathways during the early stages of tumour development. Dependence on these pathways for cell proliferation or survival allows them to acquire genetic mutations in the same pathways, providing the cell with selective advantages that promote tumour progression. Strategies to reverse epigenetic gene silencing might therefore be useful in cancer prevention and therapy.” –
“Classically, cancer has been viewed as a set of diseases that are driven by progressive genetic abnormalities that include mutations in tumour-suppressor genes chromosomal abnormalities1–5. However, it is apparent that cancer is also a disease that is driven by ‘epigenetic changes’ — patterns of altered gene expression that are mediated by mechanisms that do not affect the primary DNA sequence6–9. These epigenetic alterations occur within a larger context of extensive alterations to chromatin in neoplastic cells in comparison with the normal cells from which they are derived. These involve both losses and gains of DNA methylation as well as altered patterns of histone modifications6,8–11. Although the molecular determinants that underlie these types of chromatin change in tumour cells are only beginning to be elucidated, the best understood component is the transcriptional repression of a growing list of tumoursuppressor and candidate tumour-suppressor genes. This suppression is associated with abnormal methylation of DNA at certain CpG islands that often lie in the promoter regions of these genes6,8,9. By this mechanism of ‘silencing’, the expression of these tumour-suppressor genes in the cancer cell can be reduced or eliminated as an alternative mechanism to genetic mutation6,8,9(ref)”
Re-activating silenced genes – The “Lazarus Trick”
All of the epigenetically silenced genes in the HCT116 colon cancer can be re-activated with DNA demethylating agents or by disrupting genes that encode for DNA methyltransferases (Ex: DNMT1). Reactivating the epigenetically silenced genes can result in either cancer cell apoptosis, cancer cell senescence, or just a reduction in proliferation rates. One of the most common pathways where both DNA mutations and epigenetic silencing work synergistically to promote cancer is in the Wnt signaling pathway. This will be discussed below.
4. Wnt signaling pathway is over-expressed in cancer, due to the epigenetic silencing of Wnt antagonists
Relevance to exosomes: the Wnt pathway is also involved with exosome secretion
In the colon cancer cell line HCT116, both DNA mutations and epigenetic silencing work together to activate this pathway, which results in what is called “constitutive Wnt signaling”. This means that Wnt signaling is no longer “under control” and stays “on” all the time. In
HCT116, four members of the secreted frizzled-related gene family are epigenetically silenced (SFRP1, SFRP2, SFRP4, and SFRP5). These code for Wnt antagonists that prevent over-activation of Wnt signaling. In addition to these epigenetically silenced genes, the genes that encodes for beta-catenin synthesis (CTNNB1) are mutated, resulting in the “constitutive activation” of beta-catenin synthesis. As a result, both DNA mutations and epigenetic silencing of genes contributes to the over-activation of Wnt signaling in colon cancer.
The following diagram illustrates how “addiction to the Wnt signaling pathway” occurs via gene silencing.

Reference: Epigenetic gene silencing in cancer – a mechanism for early oncogenic pathway addiction?
“Figure 1 | Addiction to the Wnt signalling pathway through gene-silencing events. a | In normal colon epithelial cells, secreted frizzled-related proteins (SFRPs) function as antagonists of Wnt signalling by competing with Wnt proteins for binding to their receptor, Frizzled (FRZ). Expression of SFRPs is therefore the epigenetic gatekeeper step. When Wnt signalling is inactive, the adenomatosis polyposis coli (APC) complex phosphorylates β-catenin, leading to its degradation. This prevents the accumulation of nuclear β-catenin and therefore its ability to engage its transcription factor partners (TGF), which results in the differentiation and homeostasis of colon epithelial cells. Expression of APC is therefore a genetic gatekeeper step. b | When SFRP expression is lost, through epigenetic silencing of the gene that encodes it (loss of the epigenetic gatekeeper), Wnt signalling becomes activated through the receptor FRZ. This Wnt signalling potentially inactivates the APC complex (loss of the genetic gatekeeper), allowing β-catenin to accumulate in the cytoplasm and eventually in the nucleus. In the nucleus, β-catenin activates transcription of genes such as MYC, cyclin D and other genes whose products promote cell proliferation and survival rather than differentiation. This results in the expansion of colon epithelial stem and progenitor cells and formation of atypical crypt foci (ACF). c | Persistent activation of the Wnt pathway allows mutations to occur in other pathway components, such as those that permanently disable the APC complex and promote nuclear accumulation of β-catenin (loss of the genetic gatekeeper, as indicated by the bold cross). These cells are selected for because of their survival and proliferative advantages. This combination of epigentic and genetic events fully activates the Wnt pathway to promote tumour progression. Without the epigenetic events that silence the SFRP genes, mutations that disrupt the APC complex might not be sufficient to promote tumorigenesis or tumour progression.”
As can be seen above, the epigenetic silencing of secreted frizzled-related proteins (SFRPs) result in unopposed binding of Wnt proteins to the cell surface receptor, Frizzled (FRZ). Unopposed Wnt signaling results in the inability of the APC complex to adequately phosphorylate beta-catenin, which normally would result in the degradation of beta-cateinin. As a result, beta-catenin is not adequately phosphorylated and can migrate into the nucleus to turn on genes such as the oncogene MYC, cyclin D, and other genes that result in cell proliferation and survival, rather than cell differentiation and senescence. This results in the pre-malignant atypical crypt foci (ACF) to develop. Persistent activation of the Wnt pathway then result in the permanent disabling of the APC complex. As a consequence, no beta-catenin phosphorylation and degradation occurs, and now the atypical crypt foci (ACF) turns into an invasive colon cancer.
Conclusion: Without epigenetic silencing of the SFRP genes, this cascade of events may never occur. Again this shows how the prevention of epigenetic silencing may be the best cancer prevention strategy.
5. Wnt signaling can be down-regulated by exosome secretion
“Good exsomes” can export beta-catenin => down regulate Wnt signaling
There is some “good news” in this “bad news story”. Specifically, tetraspanins are plasma membrane proteins that are also found on exosomes. Two tetraspanins, CD9 and CD82, are suppressors of tumor metastasis. Expression of these two tetraspanins by a cell strongly suppresses beta-catenin signaling in cancer cells. When CD9 and CD82 are expressed, beta-catenin levels are dramatically reduced within a cancer cell. CD9 and CD82 expression induces
beta-catenin export via exosomes. In this manner, the exosomes are “transporting beta-catenin away in garbage bags”. CD82 fails to export beta-catenin in cells with low expression of E-cadherin. E-cadherin expression is up regulated by Vitamin D. This may be one of the methods by which Vitamin D can prevent the migration of cancer by increasing CD82 and the export of beta-catenin by “good exosomes”. Here is a diagram of how this works:

Reference and image source: Exosome release of β-catenin: a novel mechanism that antagonizes Wnt signaling
“Effects of CD9 on Wnt/β-catenin signaling and exosome release in vivo. (A) Reduction of BMDC-derived exosomes from CD9 knockout mice. Exosomes were purified from BMDCs derived from CD9 wild-type and knockout mice as described in Materials and methods and analyzed by Western blotting using the indicated antibodies. BMDC lysates were immunoblotted with anti-CD9 as shown in the bottom panel. (B) CD82 expression inhibits β-catenin signaling and facilitates the exosome release of β-catenin in PC3 cells. PC3 cells were transiently cotransfected with the indicated plasmids and with the TOPflash and theRenilla luciferase vectors. Activity of the β-catenin signaling pathway was quantified by measuring relative firefly luciferase activity units (RLUs) normalized toRenilla luciferase and expressed as fold change in the luciferase signal (top panel). In the bottom panel, Western blots of total lysates and exosome fractions were purified from PC3 cells transiently transfected with indicated plasmids. CD82 expression increased the exosome release of β-catenin by a factor of 3. (C) A novel role for tetraspanins in the regulation of Wnt/β-catenin signaling. At steady state, tetraspanins are organized in a signaling complex with E-cadherin at the plasma membrane. This signaling complex, including tetraspanins, E-cadherin, and β-catenin, is internalized and delivered to early endosomes. Exosome biogenesis begins with outward vesicle budding at the limiting membrane of endosomes, generating intraluminal vesicles (ILVs). These exosome-containing endosomes eventually mature into late endosomes, also known as mutivesicular bodies (MVBs). These MVBs then fuse with plasma membrane and release their intraluminal vesicles, referred to as exosomes, which contain β-catenin. The exosome targeting of β-catenin causes a reduction in the intracellular pool of β-catenin and therefore reduces Wnt/β-catenin signaling.”
Conclusion: There is a “good side” of exosomes with cancer cells. In the prescience of Vitamin D, which induces the expression of E-cadherin, the CD9 and CD82 tetraspanins can act as “tumor suppressors” by exporting beta-catenin out of the cell. With the reduction of beta-catenin levels, cell proliferation goes down. This is one mechanism by which Vitamin D can reduce the risk of death from cancer by 75%.
THE DARK SIDE OF EXOSOMES – Intracrine, Paracrine, Autocrine, and Juxtacrine mechanisms of Exosomes and their role in Viral-mediated disease, Cancer, and Aging (Senescence)
6. Rab27a and Rab27b are involved with exosome secretion –
Rab27b is associated with a poor prognosis and lymph node invasion in breast cancer, due to the secretion of tumor-derived exosomes that allow for cancer cell migration.
Several cancers have been shown to express the Rab27b small GTPase. From basic science studies, this protein is known to be involved with exosome secretion. Clinical studies suggest that Rab27b breast cancer cell growth and invasion and hepatocellular cancer growth and invasion. Here is how this works. Five GTPases are involved with exosome secretion.
Two of these, Rab27a and Rab27b. Rab27b controls multi-vesicle docking at the plasma membrane. If Rab27a is silenced by RNA interference, the size of the multi-vesicles is increased. If Rab27b is silenced by RNA interference, the multi-vesicles are redistributed along the perinuclear membrane. In other words, the exosomes cannot migrate to the plasma membrane if there is no Rab27b.

Image source “Figure 1. Schematic of exosome secretion in a cancer cell model. Exosomes are secreted via a constitutive pathway involving the Trans-Golgi Network and/or inducible pathways, which can be activated by a p53-mediated response to DNA damage or Rab GTPases. Characteristic protein and RNA components are shown for a canonical exosome. Exosomes released from cancer cells can be taken up by neighboring cells and are capable of inducing pathways involved in cancer initiation and progression.”
Breast cancer, Rab27b, and lymph node metastasis
In clinical studies, the expression of Rab27b by ER positive breast cancer cells is associated with a low degree of cellular differentiation and the occurrence of early lymph node metastasis. Specifically, the Rab27b staining score has a very high correlation with lymph node metastasis with an odds ratio of 10.1! This and other works suggests that Rab27b mediated exosome release regulates breast cancer cell growth and invasion and is a good biomarker for lymph node metastasis in ER positive breast cancer.
Hepatocellular cancer, Rab27a, Rab27b, and cancer prognosis
Hepatocellular cancer also correlates with these exosome GTPases. A study out of China showed that hepatocellular cancers differentially expressed both Rab27a and Rab27b. Both were associated with a higher risk of lymph node metastasis, a statistically significant poorer prognosis and a higher risk of death. Specifically, the hepatocellular cancers that expressed Rab27a had a 3.36-fold higher risk of death and the cancers that expressed Rab27b had a 3.37-fold higher risk of death.
· The secretory small GTPase Rab27B as a marker for breast cancer progression
· Differential expression of Rab27A/B correlates with clinical outcome in hepatocellular carcinoma
· Rab27a and Rab27b control different steps of the exosome secretion pathway
Conclusion: The association of cancer expression of the Rab27a and Rab27b GTPases that are involved with exosome secretion is strong evidence that cancers which can secrete exosomes can migrate easier and metastisize faster. This is part of the “Dark Side of exosomes.”
7. Exosomes from normal cells can “talk” to cancer cells –
As a result of this “talking”, the cancer cells can migrate.
This is another aspect of the sobering reality that I was referring to as the “Dark Side of Exosomes”. Specifically, two papers have come out in the past 9 months that show how normal fibroblasts can secrete exosomes and adipose derived mesenchymal cells secrete exosomes that can make breast cancer grow. Here is a summary:
Fibroblast exosomes and Breast Cancer
Researchers from the University of Toronto came out with a paper last year that shows how fibroblast exosomes can activate breast cancer cells via Wnt planar cell polarity (PCP) signaling (i.e. Wnt11). Co-injectin of breast cancer cells and fibroblasts dramatically enhance metastasis that is dependent on Wnt-planar cell polarity signaling. More specifically, exosomes from fibroblast have “autocrine Wnt11” on their surface. Breast cancer cells tether these Wnt11 positive exosomes which activates invasion and metastasis. Here is a diagram on how this works:

Adipose-derived mesenchymal stem cell exosomes and breast cancer
In June of 2013, researchers from Peking Union Medical College showed that the conditioned media from adipose-derived human mesenchymal stem cells can secrete exosomes that will make human breast cancer cells migrate. The breast cancer cells endocytosed the exosomes and as a result, several pathways were upregulated. One of the pathways upregulated was the Wnt signaling pathway. This suggests that mesenchymal stem cell exosomes can create a “permissive environment” for cancer cells to migrate.
Reference: Exosomes from human adipose-derived mesenchymal stem cells promote migration through Wnt signaling pathway in a breast cancer cell model “ — Here, we show that MSCs secreted 40–100 nm particles, which have the typical characteristics of exosomes, and these MSC-derived exosomes promoted migration of the breast cancer cell line MCF7. Global gene expression profiling revealed that several cancer-related signaling pathways were upregulated after exosome treatment in MCF7, and the Wnt signaling pathway was further confirmed to be activated. Our findings demonstrated a new mechanism through which MSC-CM may contribute to tumor cell migration.”
8. Exosomes from cancer cells can “talk” to normal cells
This can result in a favorable microenvironment for cancer cells, resulting in the creation of a “premetastatic niche”.
More articles have been written about tumor exosomes than any other aspect of exosomes. In pre-malignant tumors where p53 is not mutated, there is robust exosome synthesis and secretion. (This is not true for cancers where p53 is mutated, however). These tumor-derived exosomes have been observed for many different types of cancers, including ovarian cancer and, breast cancer,
Additional references:
- Exosomes from ovarian cancer cells induce adipose tissue-derived mesenchymal stem cells to acquire the physical and functional characteristics of tumor-supporting myofibroblasts.
- Exosomes from breast cancer cells can convert adipose tissue-derived mesenchymal stem cells into myofibroblast-like cells.
- Exosomes derived from human bone marrow mesenchymal stem cells promote tumor growth in vivo.
The “bright side” of tumor exosomes
The body’s immune system can recognize tumor exosomes, in some instances and mount an immune response against the cancer. This is the “bright side” of tumor exosomes”.
The “dark sides” oftumor exosomes
Unfortunately, there are many more “dark sides of tumor exosomes”.
Cancer-derived exosomes can induce the expression of VEGF by nearby endothelial cells by the uptake of the cancer exosomes by the endothelial cells. The cancer exosomes have activated EGFR on them. Cancer-dervied exosomes have been shown to make cancers grow faster. Thus, this may be a cell-to-cell signaling mechanism between the exosome-secreting tumor cells and their surrounding milleu of other cancer cells (some of which may not be able to make exosomes due to p53 mutations). In this aspect, exosomes may function similarly to how bacterial quorum sensing functions, creating a cancer cell bystander effect.
Cancers can use exosomes to evade the immune system by several methods:
1. This can be mediated byTGFbeta down-regulation of NGK2D, an activating receptor for NK, NKT, CD8, and other T cells.
2. Cancer exosomes can also impair lymphocyte response to IL-2, making NK cell therapy for cancer less effective.
3. Cancer exosomes can also express death ligands and induce apoptosis of activated T cells.
Cancers can send microRNAs to other cells. Over 300 different microRNA have been found in tumor exosomes. Since early tumors are more likely to have intact, functional p53 pathways, the use of exosomes as biomarkers for the early detection of tumors is much more promising than for late, metastatic tumors where p53 is more likely to be inactivated.
References:
- The Roles of Tumor-Derived Exosomes in Cancer Pathogenesis
- Purification, Characterization and Biological Significance of Tumor-derived Exosomes
- MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer
- Human Tumor-Derived Exosomes Down-Modulate NKG2D Expression1
- Human Tumor-Derived Exosomes Selectively Impair Lymphocyte Responses to Interleukin-2
- Circulating MicroRNA Signatures of Tumor-Derived Exosomes for Early Diagnosis of Non–Small-Cell Lung Cancer
- Tumor exosomes expressing Fas ligand mediate CD8+ T-cell apoptosis
- Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR
- Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming
- Human tumor-derived vs dendritic cell-derived exosomes have distinct biologic roles and molecular profiles
- Surface anchorage of superantigen SEA promotes induction of specific antitumor immune response by tumor-derived exosomes
- Tumour-derived exosomes or microvesicles: another mechanism of tumour escape from the host immune system?

9. Prostate Cancer, Radiation-induced senescence, and exosome synthesis
Radiation induced senescence of prostate cancer cells and replicative senescence of prostate cancer cells can both induce exosome synthesis and release, if p53 is not mutated.
Three primary responses mediated by p53 occur with radiation: Cell cycle arrest, Apoptosis, or Cellular Senescence.
Cellular Senescence is a form of cell cycle arrest that has often been referred to as “permanent cell cycle arrest”. Recent evidence suggests that the most common of these three primary responses to radiation is Cellular Senescence. The development of cellular senescence following radiation is not “instant” and takes about 4 days to occur. Cellular senescence also occurs with aging and can be due to replicative senescence due to telomeres becoming too short. This is why human cells have a “Hayflick limit”, where they will only divide for a certain number of times and then will no longer divide. This is thought to be due to the “end replication problem” where telomeres cannot be replicated to their full length during the S-phase of DNA replication, since the enzyme Telomerease requires a template to synthesize telomeric DNA.
Prostate cancer is one of the cancers most closely associated with old age and cellular senescence to start with. As a result, there are many senescent cells in the prostate even in the absence of prostate cancer. Recent studies have shown that radiated prostate cancer cells that have undergone senescence have a robust secretion of exosomes. After radiation, there is an “immediate exosome synths” and a “delayed exosome synthesis”. (In reality, the correct terminology for the “immediate exosome release” is probably microparticles or apoptotic bodies). With the “immediate exosome biogenesis,” exosomes are released by “outward budding” of the plasma membrane. This is the classic description for the release of apoptotic bodies from cells that are dying and does not require any “protein sorting complex”, such as the ESCRT (Endosomal Sorting Complex Required for Transport).
In contrast, the “delayed exosome biogenesis” occurs 4 days after radiation and starts with outward vesicle formation from the intracellular endosomal compartment, called the “limiting membrane”. The budding of the “limiting membrane” produces mutlvesicular bodies (MVEs) with many exosomes inside the MVEs, like “Russian Dolls”. When this form of exosome synthesis was inhibited by siRNA “knock down” of one of the proteins involved (Tsg101), a 65% reduction in exosome release was seen following radiation of the prostate cancer cells.
In a separate experiment, a study was done to see if increasing p53 would increase exosome production. An inhibitor of MDM2 called Nutlin-3, a small molecule inhibitor of this pathway that disrupts the MDM2 negative feedback loop of p53, was added to the prostate cancer cells prior to radiation. This “primed” the cells by increasing the amount of p53 present.
Since p53 was not degraded by the MDM2 ubiquitinylation. Then the prostate cancer cells were radiated with only 2 Gy, rather than the 4Gy that was given in the above experiments. This dramatically increased the number of cells that died due to apoptosis or cell necrosis. The same experiment was then done with the 4Gy dose, 30 minutes after Nutlin-3 introduction.
This increased exosome production by 2-fold.
Then, prostate cancer cells that had reached cellular senescence by replicative senescence were examined for exosome release. Compared to the non-senescent cells, there was a 15-fold increase in the synthesis and secretion of exosomes from the senescent cells. This suggests that both radiation induced senescence and replicative induced senescence increases exosome synthesis and secretion.
Conclusion: Most of the exosomes produced by radiated prostate cancer cells that had become senescent were the exosomes involved with the “delayed exosome biogenesis” pathway which involved the formation of MVEs from the limiting membrane of the exosomal compartment. This activation was totally dependent on p53 activation and was not seen in cancer cells where the p53 pathway was non-functional. Both radiation induced senescence and replication induced senescence increases p53 mediated exosome synthesis and secretion. Thus it appears that cellular senescence is a state that is dominated by exosome secretion. This is totally logical, since only cells with a functional p53 pathway can undergo cellular senescence.
In short, aging is accompanied by a robust secretion of exosomes. These exosomes are not “good exosomes” and probably mediate much of the bad effects of senescent cells, such as chronic inflammation and the conversion of premalignant cells into malignant cells. This is another manifestation of the “dark side of exosomes.”
Reference: Senescence-Associated Exosome Release from Human Prostate Cancer Cells

Related to this is the signals of mitochondria to be recycled (Autophagy). I still believe that mitochondria will prove to be a key if not the key to a age firewall. Eric
Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nature Cell Biology, 2013; DOI: 10.1038/ncb2837
Eric35001
You may well be right with respect to the importance of targeting mitophagy as part of an anti-aging firewall. Thanks for the citation on cardiolipin as a eat-me molecule for mitochondria. It is interesting that anti-cardiolipin antibodies are present in many disease processes, particularly autoimmune diseases. I wonder if that contributes to limiting mitophagy in disease-infected cells.
Vince
I notice your article occasionally, very useful for my grandmother who is sick in recently. I’ll bookmark your site. Thx.