
By James P Watson with editorial and some content contribution by Vince Giuliano
This is the second blog entry in a three-part series concerned with transposable DNA entries (TEs). The Part 1 entry focused on basics of what TEs are, and why they are important for us who are concerned with health and aging. This Part 2 entry is the main course of the series, providing the “meat and potatoes” of technical content. Twenty one key aspects of TEs are discussed here. Part 3 of the series TEs and an emerging “Grand Unified Theory” of biology and aging outlines the key role of TEs in making evolution possible and how TEs fits into an emerging unified theory of how all life and aging works.
1. There are Over 2 Million Copy Machines in your Genes, but only 80-100 still work (they are all LINE-1s)
These TE “copy machines” drove evolution for 150 million years, but have largely become non-functional in humans. However, those that are working can still copy themselves (LINE-1s) or copy other transposable elements that were never able to copy themselves (i.e. were parasitic transposable elements). The Alu and SVA transposable elements are the “parasitic TEs”.
Here are the details.
HERVs (Human endogenous retroviruses)
There are 98,000 fragments of leftover “fossil viruses” in our genomes (HERVs) but none are capable of “self-copying” themselves. In rodents, however, there are many mouse endogenous retroviral elements (MERVs) that are “active” and capable of retrotransposition. This is one of the major genetic differences between “mice and men” and may partially explain why TE insertional mutagenesis in rodents is 100-fold higher than in humans. Likewise in mice several MERVs are very similar looking to exogenous, infections mouse retroviruses (This is why many viruses cause tumors in mice and rats). The fact that LTR-retrotransposons cannot copy themselves in humans is surprising, since HERVs make up 8-9% of the human genome (in terms of DNA base pairs). When transposable elements cannot copy themselves, they are referred to as being “non-autonomous”, which HERVs are in humans (but not rodents). This difference between humans and rodents may be a major reason why we live so much longer than rodents.
So, largely because of active TEs, mice are more open to evolution than we are and they get a lot more cancers.
Although the HERVs in us are non-autonomous, this does not mean that they are completely benign. Some copies with ORFs can still be transcribed (into RNA) and even fewer copies have their RNA translated into env proteins, gag proteins, and 2 small proteins that are very important in cancer, called Rec and Np9.
The 2015 publication Human Endogenous Retrovirus Type K (HERV-K) Particles Package and Transmit HERV-K-Related Sequences relates how a retrovirus can spread itself to other cells, although not necessarily creating any pathology: “Retroviruses invaded the genome of human ancestors over the course of millions of years, yet these viruses generally have been inactivated during evolution, with only remnants of these infectious sequences remaining in the human genome. One of these viruses, termed HK2, still is capable of producing virus particles, although these particles have been regarded as being noninfectious. Using a genetic probe derived from HK2, we have discovered that HK2 viruses produced in modern humans can package HK2 sequences and transmit them to various other cells. Furthermore, the genetic sequences packaged in HK2 undergo reverse transcription. The transmitted probe circularized in the cell and failed to integrate into the cellular genome. These findings suggest that modern HK2 viruses can package viral RNA and transmit it to other cells. Contrary to previous views, we provide evidence of an extracellular viral phase of modern HK2 viruses. We have no evidence of sustained, spreading infection.”
One unique “fossil virus” called HERV-18 can also produce a “superantigen” which makes your T cells very “angry”. The expression of HERV-18’s superantigen may be why the exogenous Eptstein Barr Virus (EBV) is able to establish a long-term infection status see in humans. For the HERV sequence to be transcribed, the fossil viral DNA must have an open reading frame (ORF). 3 families of HERVs still have ORFs (HERV-K, HERV-FRD, and HERV-W). As a result, proteins from these three classes of HERVs have been detected, including env, gag, rec, Np9, and the HERV-K18 super antigen(ref). The last member of the HERV family of viruses to infect the human genome was the HERV-K(HML-2) subfamily, which makes up 1% of all HERVs. These did infect the human genome since we “split” from chimpanzees. Some copies of HERV-K(HML-2) have been active (i.e. transfecting the human genome) in the past few hundred thousand years. HERVs with ORFs use RNA polymerase II (Pol II) to transcribe their DNA.
Role of LTRs in HERVs
However, the most important role of HERVs today has nothing to do with transcription or translation. Instead, it is the role of the “fossil promoter”, called the “LTR” (long terminal repeat), which can function as an “alternative promoter” to activate nearby human genes which is the most important function of HERVs in our DNA. These LTRs are used in embryonic stem cells to activate “pluripotency programs”, since the LTR promoter has a binding site for OCT4. In the placenta, the envprotein of the HERV-W “fossil virus” is used to trigger cell differentiation. In many diseases, the LTRs are used to activate specific genes, since there are transcription factor binding sites for so many human transcription factors. In both mice and humans, LTRs play an important role in disease pathogenesis.
However, since LTRs are no longer mobile in humans, no insertional mutagenesis events have been discovered that were due to HERV mobility (Maksakova, 2006). If HERVs play a role in cancer, it is likely to be via their ability to function as “alternative promoters” for oncogenes or other genes that dysregulate signaling pathways that prevent cancer (i.e. apoptosis, autophagy, DNA repair, etc.).
LTRs and inflammation
LTRs that function as “alternative promoters” may play a major role in inflammaging, since there are many inflammatory transcription factors that can bind to HERV LTRs and activate gene expression. The link between inflammation, LTRs, and aging has not been clearly proven, however.
LINEs – “Seventeen per cent of the human genome is comprised of ‘parasitic’, repeated DNA segments called LINE-1 elements. Some of these, occasionally, copy themselves and move to new locations in the genome. More rarely, they will aberrantly pick up a piece of adjacent, non-LINE-1 DNA and copy that to a new site: this is known as LINE-1 transduction(ref)”. There are 850,000 fragments of LINEs left over from evolution, although only about 7,000 of these are intact. Evolutionary speaking, LINE-1s are very old, first showing up in yeast (C. albicans) about 150 million years ago. Of the intact copies of LINE-1, only between 80-100 are capable of “self-copying”. This means LINE-1s have DNA that codes for all the enzymes that allow them to be transcribed into RNA, then be converted back into DNA copies (i.e. reverse transcriptase), and then reintegrate into the genome.
Rodents and humans both have functional copies of LINE-1, so these LINEs are called “autonomous retrotransposons”. All of the 80-100 functioning “self copy machines” within the human genome are from the LINE-1 (L1) family. L1s can also copy other TEs, such as Alu or SVA elements. These two types of TEs are non-autonomous and are completely dependent on the L1 copy machines to be duplicated, then re-inserted into new sites in the human genome. In addition to copying Alu elements and SVA elements, the functioning L1s can also copy nearby human genes. This is why as we age, humans become a “genomic mosaic”, where not all of the cells have the same copy number of genes, such as the Amyloid Precursor Protein (APP) gene in the brain. Genome mosaicism is another fascinating topic that deserves a lot of attention, since it may be one of the major causes of sporadic Alzheimer’s disease. See Item 14. below
What good are HERVs and LINE-1A today?
Whereas the beneficial, functional role of HERVs today is mostly due to the “repurposing” of the LTR as an “alternative promoter,” the main beneficial, functional role of LINEs in human cells is not as clear. Although they clearly played a beneficial role in evolution, today they appear to primarily play a role in producing harmful mutations and disease. For instance, in normal cells, LINEs are very poorly expressed, whereas in cancer cells, L1s are abundantly expressed and decreasing LINE-1 activity in cancer cells reduces cancer cell proliferation and promotes cancer cell differentiation (thereby stopping proliferation). This diagram illustrates how LINE-1s can spread DNA around in lung cancer cells. One out of every four Line-1 mutations is a transduction, copying of adjacent non-LINE-1 DNA.
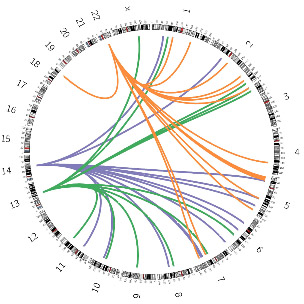
Image source ” In a lung cancer genome, three LINE-1 copies located at human chromosomes 22, 14, and 13 spread non-repetitive DNA by transduction. [DOI: 10.1126/science.1251343]”
There are at least a dozen other diseases besides cancer that have been specifically linked to L1 insertions into cells. This includes choroideremia, chronic granulomatous disease, X-linked Duchenne muscular dystrophy, Hemophilia A and B, X-linked retinitis pigments, Coffin-Lowry disease, Fukuyama-type congenital muscular dystrophy, beta-thalasemia, and Pyruvate dehydrogenase deficiency. For the above reasons, a great amount of attention has been made towards understanding how to silence LINE-1 expression in human cells.
Alu SINE – Alu elements are “copiers that borrow their copy machine“. The Alu transposable element was named for the Alu Arthrobacter luteus restriction endonuclease enzyme. Alu is part of the family of Short Interspersed Nuclear Elements (SINEs) that is evolutionarily derived from a mutated copy of a small cytoplasmic 7SL RNA, which is a part of the signal recognition particle. Alu is a primate-specific transposable element.
Unlike HERVs or LINEs, none of the Alu DNA sequence codes for proteins. This makes it a non-autonomous TE, which must use the L1 “copy machine” to duplicate itself. Despite those limitations, Alu elements have been twice as successful as L1 TEs in producing copies of themselves (1.4-2 million vs 850,000 copies), even though LINE-1 is a much older TE, in terms of evolution (150 million years vs 65-80 million years). This makes the Alu element “ the most successful copier” in mammalian history, based on copy number. “How did primates get to begat so many other primates eventually begatting us?” Was it due to the Alus? What is clear is that Alu elements have developed many important, indispensable molecular roles in primate evolution (Ex: RNA editing, gene expression regulation, etc.), especially in the evolution of the primate brain.
The “evolutionary success in copying rate” of Alu has been attributed to “retrotransposition efficiency”, post-insertional evolutionary selection, or both. The retrotransposition efficiency of Alu elements may be due in part to the fact that they only require ORF2 in the LINE-1 copy genes, whereas LINE-1 copying requires both ORF1 and ORF2 to successfully undergo retrotranspostion. There are more L1s in the human genome with intact ORF2s than there are L1 copies with intact ORF1s (i.e. the old, “partially broken copy machines” can still copy Alu elements).
Alu elements also have deleterious effects in humans, with mutagenic effects and a role in diseases. Alu element copying can introduce insertions and deletions in genes, and have been shown to cause insertions in 33 genes, producing many more diseases than associated with LINE-1 insertions. The Alu insertion-induced diseases include breast cancer, Hemophilia A and B, Muckle-Wells syndrome, chronic hemolytic anemia, cholinesterase deficiency, type 1 antithrombin deficiency, cystic fibrosis, mucolipidosis type II, Walker Warburg syndrome, Apert syndrome, Ewing’s sarcoma, familial hypercholesterolemia, hemophilia, neurofibromatosis, diabetes type II, and may also play a role in Alzheimer’s disease, lung cancer, and gastric cancer.
This Table showing various possible Alu LI and SVA insertions and the associated diseases is from the reference: (Belancio, et.al, 2008) Mammalian non-LTR retrotransposons: For better or worse, in sickness and in health.

An important role for Alu elements is in the regulation (or dysregulation) of microRNAs. Recently it was shown that as much as 20% of all known human microRNAs are driven by Alu Pol III promoters. Dysregulation of microRNAs is a ubiquitous feature of cancer.
SVAs – SVAs are chimeras that are “hominid-specific.” SVAs are an uncommon type of TE found in humans that are “very young” in terms of evolution. Less than 3,000 copies of SVAs exist in the human genome and they appear to have played a recent role in the evolution of our ancestors. SVAs started integrating into the hominid genome about 25 million years ago and are not found in old-world monkeys. They may have been key in giving homonid evolution a final boost and putting us over the top as homo sapiens.
From their DNA sequence, SVAs look like a chimera, a hybrid of several structures, which makes them difficult to classify. Some call them a successful “pseudogene”, whereas others classify them as a retrotransposon (i.e. a TE). They are part of the SINE family and are non-autonomous, so they have to use the L1 “self-copy machine” to replicate and reintegrate. The acronym SVA stands for SINE VNTR Alu, since this TE has a Variable Number of GC-rich Tandem Repeats (VNTR) and a sequence that is similar to two anti-sense Alu fragments. In addition, the SVA also has a DNA sequence that is very similar to the HERV-K env gene and the right LTR sequence of the HERV-K10 “fossil virus”.
Although it is clear that SVAs played a role in hominid evolution, no current beneficial, functional role for SVAs has been identified in humans. They have been found to induce insertional mutations and deletions of genomic material, playing a role in at least 7 diseases (leukemia, neurofibromatosis 2, X-linked agammaglobulinemia, Hereditary elliptocytosis and pyropoikilocytosis, X-linked dystonia-parkinsonism, Autosomal Recessive Hypercholesterolemia, and Fukayama-type muscular dystrophy,
Reference: (Hancks, Kazaian, 2010). 2010 SVA retrotransposons: Evolution and genetic instability “SVAs are currently active and may impact the host through a variety of mechanisms including insertional mutagenesis, exon shuffling, alternative splicing, and the generation of differentially methylated regions (DMR).”
Here is another depiction of the different kinds of TEs with a legend commenting on their characteristics:
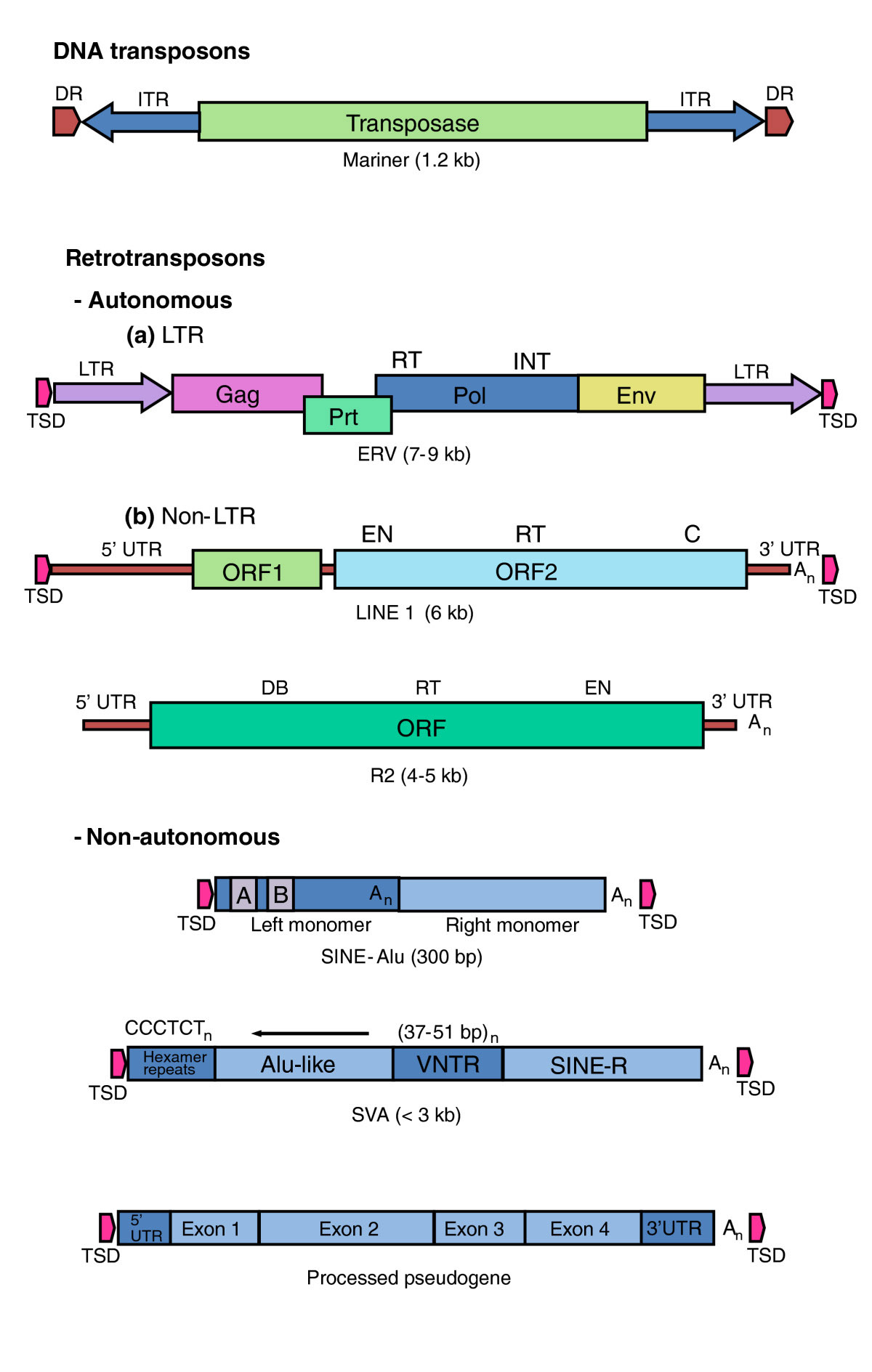
Image and legend source “Types of transposable element. DNA transposons such as the Mariner-like elements in mammalian genomes are inactive relics of mobile DNAs that transpose directly. They encode a transposase activity that mediates their excision from, and integration into, DNA. Retrotransposons transpose via an RNA intermediate that is converted by a reverse transcriptase (RT) into a DNA that is integrated into the host-cell genome. Retrotransposons that contain many (but not all) of the activities necessary for their mobility are called autonomous. They comprise the endogenous retroviruses (ERVs), which are characterized by (a) long terminal repeats (LTRs) and (b) the LINEs, which are non-LTR retrotransposons. Of the LINES, only the L1 group is known to be actively mobile in eutherian mammals. R2 is a site-specific element that is active in insects and is the element studied by Eickbush and Eickbush [5]. Non-autonomous elements, such as Alu and SVAs, are dependent on L1 for their mobility. Processed pseudogenes derive from spliced RNAs that are copied and inserted in the genome by the L1s. Mariner: DR, direct repeat (in the host-cell DNA); ITR, inverted terminal repeat. ERV: TSD, target-site duplication (in the host cell DNA); LTR, long terminal repeat; Pol, RNA-dependent DNA polymerase with a reverse transcriptase (RT) domain and an integrase (INT) domain; Env, Gag and Prt encode other proteins required for the virus life cycle. LINE: EN, endonuclease domain; RT, reverse transcriptase domain; C, zinc knuckle domain; DB, DNA binding; An, poly(A). SINE: A/B, A- and B-box PolIII promoter.”
Summary: Alus are the most successful “copiers”, and also have been linked to the highest number of diseases caused by insertional mutations of specific genes. SVAs are the most recent “copiers,” are the least common, and have been linked to the fewest number of diseases caused by insertional mutagenesis of specific genes. LTR retrotransposons are no longer active in the human genome, whereas they are still mobile in rodents (mice and rats). This means they play no role in insertional mutagenesis in cancer in humans, whereas ERVs may play a major role of insertional mutagenesis in rodents.
LINE-1s are the only class of TEs with copy machines that still work in humans, with only 80-100 copy machines still running. However there are another 100 or so where only ORF2 works. Only ORF2 is needed to copy Alu elements. This may be one reason there are twice as many Alus as LINEs in humans.
Alus have played a crucial role in primate evolution and continue to play a role in human evolution, since an additional 7,000 copies of AluY have been inserted into the human genome since we “split” from our common ancestor with chimpanzees. In addition to their role in evolution and disease, Alus have clearly been genetically “repurposed” to play many vital roles in gene development, RNA editing, gene regulation, and microRNA regulation. As a race, humans owe a debt of gratitude to this 300 bp piece of DNA that has copied itself over a million times in our genomes. Without it, we would look much more like our mammalian primate and non-primate relatives.
2. Short-lived organisms are more permissive to retrotransposition, whereas long-lived organisms are resistant to retrotranspostion.
Compared to humans, mice have a 100-fold higher rate of mutations caused by retrotransposon insertions (10% vs 0.1-3%). This is the strongest line of evidence that the ability of an organism to repress TEs correlates with lifespan. 99.9% of TEs in humans are incapable of retrotransposition. This fact is the second strongest line of evidence that “mobile-capableTE expression” in the genome correlates negatively with life span.
Retrotransposable element insertions into the human genome are 20 times more numerous, per unit of DNA, when compared to retrotransposable element insertions into the fruit fly genome (Drosophila melanogaster). Despite this high rate of TE integration in humans, we have very low rates of TE activity. In fruit flies, these TEs are subject to constant turnover (new insertions of TE and elimination by natural selection, i.e. death of the organism) (Eickbush, Furano, 2002 Fruit flies and humans respond differently to retrotransposons). In fruit flies (D. melanogaster), only 12% of genomic DNA consists of transposable elements, yet more than 50% of de novo mutations arise from these transposable elements. (Eickbush, Furano, 2002)(Bartolome, et al, 2002 On the Abundance and Distribution of Transposable Elements in the Genome ofDrosophila melanogaster).
This is thought to be due to the fact that TEs are very mobile in fruit flies. In mice, 37% of genomic DNA consists of transposable elements, but only 10% of all de novo mutations in mice are due to TE activity. This reduction in the percentage of mutations due to TEs parallels the reduction in TE mobility in mice, compared to fruit flies. So there appears to be a consistent trend illustrated in this comparison of fruit flys, mice and humans.
In humans, the same trend occurs. Approximately 45% of genomic DNA in humans is comprised of TE sequences, yet it has been estimated that only 0.3% of de novo mutations in human cells are due to TE activity (Deininger, Batzer, 1999 Alu Repeats and Human Disease). Compared to mice, TEs in humans are much less mobile (Belancio, et.al., 2008) (Maksakova, et al, 2006 Retroviral Elements and Their Hosts: Insertional Mutagenesis in the Mouse Germ Line). Perhaps the most important point here, however is that LTR retrotransposons are no longer mobile in the human genome and as a consequence, they do not produce gene mutations by TE insertions. Likewise, although there are 850,000 fragments of LINEs in the human genome, only 80-100 contain functional ORF1 and ORF2 segments. As a consequence, 99.9% of the TEs in the human genome can no longer produce mutations.
So there appears to be a consistent trend illustrated in this comparison of fruit flies, mice and humans. As the organisms get more advanced and sophisticated there are larger percentages of TEs in the genomes but the TEs become much less active and capable of inducing mutations.
TE silencing mechanisms are also much more complex in human cells, with many more layers of repressive molecular mechanisms, compared to more primitive organisms. This includes histone biotinylation, histone trimethylation, histone deacetylation, DNA methylation, siRNA-mediated degradation of TEs, and PIWI-piRNA pathway degradation of TEs. These multiple layers of TE suppression at the transcriptional and post-transcriptional level is clearly an additional reason why humans have less TE activity, compared to Drosophila and other lower organisms.
Conclusion: TE activity is low in human somatic cells, compared to mice, fruit flies, nematodes, and yeast. Paradoxically, the sheer DNA content (as a percentage) of TEs does not vary directly with TE activity. Instead, there seems to be an inverse relationship that cannot be easily explained.
One possible explanation is that fewer copies of the TEs are still functional and active in humans. This is clear for the LTR retrotransposons, which are still active in rodents but no longer capable of transposition in humans. However, the more extensive silencing mechanisms in humans is more likely to explain the paradox of TE content and TE activity in humans vs fruit flys or mice.
3. Human Gene Transcription or Repression can be triggered from the anti-sense promoters of LINE-1 and from the bidirectional LTRs of old HERV sequences – This may be how non-coding genes evolved, since they are often transcribed off of the anti-sense strand of DNA.
A major flaw in the classical “Watson-Crick model” of gene expression is the widely held belief that genes are primarily regulated by transcription factors binding to classical promoter regions found near the transcription start sites of genes. Whereas this concept is valid, many sequences of DNA upstream and downstream from these canonical promoters can affect gene transcription. Specifically, the structure of the 5′-UTR region of L1 contains both a “sense promoter” (SP) and an “anti-sense promoter” (ASP). (If you need to refresh your understanding of mRNA structures, this publication explains how they are organized, the elements in them including UTRs and ORFs. and possible post-transcription regulatory functions of the UTR elements on gene expression.) These two promoters allows the LINE-1 TE to be transcribed from the SP and an upstream human gene to be transcribed from the ASP. This is likely to be an important mechanism for intergenic, long noncoding RNA genes to be transcribed. Here is a diagram illustrating this:
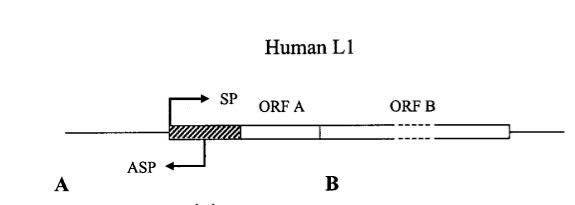
Image reference: 2002 Many Human Genes Are Transcribed from the Antisense Promoter of L1 Retrotransposon
In a query of an expressed sequence tag (EST) database of GenBank, using RepeatMasker, researchers found 23 cESTs which could be driven by L1 antisense promoters in normal cells. In tumor cells, an additional 19 cESTs were found in cell lines from teratocarcinomas, colon cancers, prostate cancers, liposarcomas, ovarian cancers, and B-cell tumors. Moreover, in many more cases, they found full length LINEs within introns of genes that allowed for the adjacent human gene to be transcribed from the anti-sense L1 promoter. Their conclusion was that although gene activation could occur from a perfectly placed anti-sense L1 promoter, in the majority of cases, the L1 anti-sense promoter down regulated the gene expression of the nearby gene by producing only a partial transcript of the gene or by silencing the entire gene.
Reference: 2002 Many Human Genes Are Transcribed from the Antisense Promoter of L1 Retrotransposon
4. TEs and inflammation
Promoter sequences from L1 and Alu elements can also be used to trigger inflammatory pathways. A recent study showed that the IFN-gamma gene can only be maximally activated with an alternative promoter, derived from Alu and LTR families of TEs.
2009 The role of transposable elements in the regulation of IFN-λ1 gene expression “Studies of virus induction of IFN-λ1 identified an essential role of IFN regulatory factors (IRF) 3 and 7, which bind to a regulatory DNA sequence near the start site of transcription. Here, we report that the proximal promoter region of the IFN-λ1 regulatory region is not sufficient for maximal gene induction in response to bacterial LPS, and we identify an essential cluster of homotypic NF-κB binding sites. Remarkably, these sites, which bind efficiently to NF-κB and function independently of the IRF3/7 binding sites, originate as transposable elements of the Alu and LTR families. We also show that depletion of the NF-κB RelA protein significantly reduces the level of the IFN-λ1 gene expression. We conclude that IFN-λ1 gene expression requires NF-κB, and we propose a model for IFN-λ1 gene regulation, in which IRF and NF-κB activate gene expression independently via spatially separated promoter elements. These observations provide insights into the independent evolution of the IFN-λ1 and IFN-β promoters and directly implicate transposable elements in the regulation of the IFN-λ1 gene by NF-κB.”
5. Bidirectional promoters and diseases
LTRs can also function as bidirectional promoters, activating transcription in both directions. The best know examples of this is the activation of expression of GSDML by an LTR promoter, which can be activated by an LTR transposon. This causes chronic myeloid leukemia and four other cancers. Another example involves the genes involved with autoimmune disease – HLA-DQB1*04 and HLA-DRB1*04. A nearby LTR sequence can function as an “alternative promoter” and activate these genes in rheumatoid arthritis. A LTR sequence near another HLA-DQ gene can activate this autoimmune gene in type 2 diabetes.
See the 1993 publication Expression of human endogenous retrovirus (HERV-K) in chronic myeloid leukemia “We have previously demonstrated the presence of a reverse transcriptase-like enzyme in retroviral particles from patients with essential thrombocythemia, polycythemia vera, and chronic myelogenous leukemia. It was subsequently shown that the human genome contains 50 copies of HERV-K. HERV-K is a human endogenous class I retroviral element that contains gag, pol, and env open reading frames. Using both reverse transcriptase-polymerase chain reaction and ribonuclease protection assays, it is demonstrated that the HERV-K pol is expressed in human blood leukocytes. The data indicates that this expression is restricted in CML white cells and is the result of gene regulation.”
References:
2010 A role for human endogenous retrovirus-K (HML-2) in rheumatoid arthritis: investigating mechanisms of pathogenesis. “The significant variation in RA patients, both serologically and transcriptionally, may be an indication that RA is an umbrella term for a number of separate disease entities, of which particular HERV polymorphisms may play a role in development.”
2009 The role of transposable elements in the regulation of IFN-λ1 gene expression
LTRs can also down-regulate nearby human genes. In Schizophrenia, a nearby LTR can down regulate the GABBR1 gene, which encodes for the GABA receptor. Male infertility can also be cause by a mutation in a gene found on the Y chromosome, AZFa, which results in a loss of function of this gene.
References:
2013 GABBR1 has a HERV-W LTR in its regulatory region – a possible implication for schizophrenia
LTRs can also result in mislocalization of a protein. Although the exact mechanism is not clear, this may involve the deletion or alteration of a nuclear localization sequence on the gene. This may be the case with certain cases of Amyotrophic lateral sclerosis (ALS), where LTR activation causes TDP-43 mislocalization to the cytoplasm, rather than the cell nucleus:
Reference: 2011 Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis
Summary: There is a small, but well-documented role for the effects of L1 promoter and LTR sequences from old HERVs on human gene expression.
Because the promoters for LINE-1 non-LTR retrotransposons and LTR retrotransposons can induce bidirectional gene expression. The sense strand usually triggers transcription of the retrotransposon, whereas the anti-sense strand promoter can initiate gene expression of an “upstream” human gene. This includes genes that can be activated by the antisense promoter, genes that can be silenced by the upstream promoter, and gene products that loose either part or all of the functionality.
6. SIRT6 “vacates” L1 promoters with aging, allowing L1 expression to occur in old age
Turning to the highly practical, SIRT6 is one of the major mechanisms of suppressing LINE-1 expression. SIRT6 ribosylates KAP1, a nuclear corepressor protein, and facilitates KAP1 interaction with heterochromatin factor 1 alpha (HP1a). This silences LINE-1 retrotransposons. SIRT6 binds to a site in the 5′-UTR region of the L1 retrotransposon, thereby inhibiting transcription of the TE.
From Wrangling Retrotransposons (2015): “Specifically, SIRT6 binds to the L1 5′ UTR, where it coordinates the assembly of heterochromatin proteins at the L1 promoter. Once assembled, this silencing machinery saturates the L1 5′ UTR and its associated chromatin with epigenetic modifications that prevent the retrotransposon from being expressed. — Without the ability to express itself, L1 cannot synthesize the parasitic machinery required to initiate its own replication. As a result, the host cell is protected from de novo mutagenesis events. Additionally, because the L1 is heterochromatinized, it has limited ability to interfere with host gene expression or to induce erroneous recombination events. When SIRT6 is deleted from mouse cells or knocked down in human cells, the L1 5′ UTR escapes heterochomatinization, resulting in massive increases in L1 transcription and de novo L1 insertions.”
Unfortunately, this does not necessarily work so well with aging. SIRT6 becomes depleted from the L1 5′ UTR in the course of aging. A consequence is the de-heterochromatinization of the L1 element allowing that element to become transcriptionally active. This has been seen in senescent human cells as well as in multiple tissues of aging mice.
A related issue is that when double-stranded DNA damage occurs, SIRT6 “runs away” like an ambulance chaser to the site of the DNA damage. When SIRT6 “runs away”, this leaves the L1 5′-UTR region “naked”, and then transcription of the two open reading frames (ORF1 and ORF2) of the LINE-1 element can occur. Unfortunately, many copies of L1 are typically produced by this. As a result, the cells either “age” and become senescent or they die. Both oxidative stress and aging trigger this molecular mechanism. Later, we mention how in aging keeping up the amount of NAD+ and maintaining a high NAD/NADH ratio can provide an adequate supply of SIRT6 and avert this.
Reference: 2014 SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age
Here is an illustration which shows how SIRT6 “relocalizes” to a site where DNA damage is occurring.
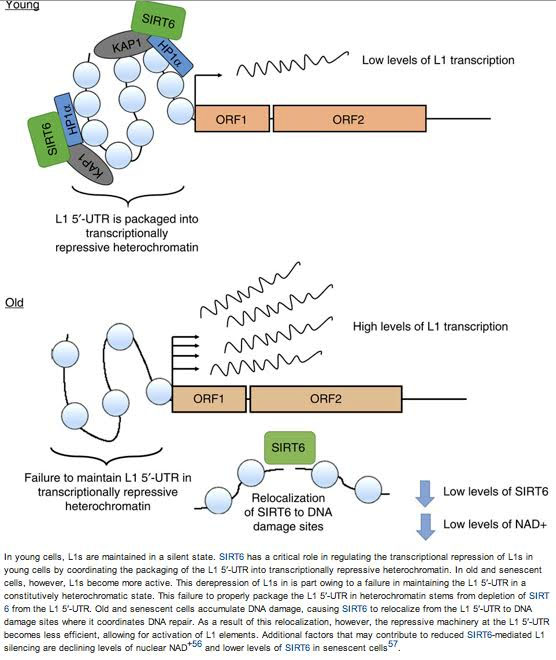
As a result, it leaves the ORF1 “naked”, allowing the L1 retrotransposon to be transcribed by RNA Polymerase. This is why “we are screwed” by low NAD levels and low NAD/NADH ratios.
Unfortunately, Resveratrol and Glaxo-Smith-Kline’s synthetic STAC activators do NOT activate SIRT6 like they do SIRT1. Neither does pterostilbene, curcumin, EGCG, and other polyphenols. If we want to have enough SIRT6 around to keep or L1 TEs silent we need to pay attention to our NAD levels and NAD/NADH ratios. You can check out our recent four-part series of blog entries regarding the NAD world (ref)(ref)(ref)( ref).
Reference 2014 SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age
Here is the on the difference between L1 expression in young cells and old cells in the brain, liver, and heart;
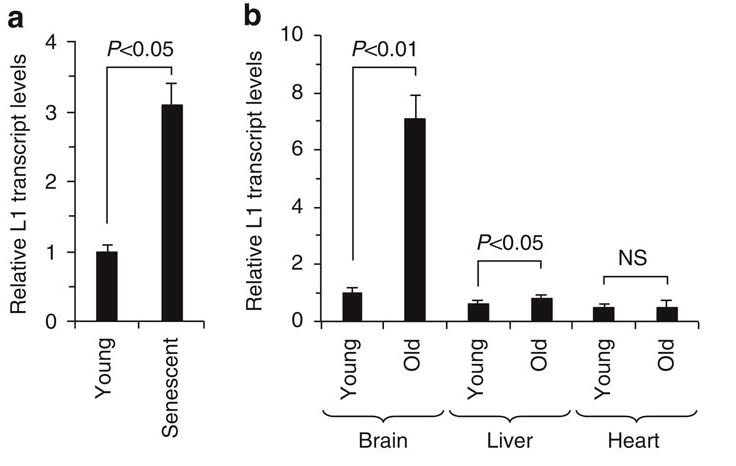
Illustration reference: 2014 SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age Fig. 4
Here is the data that shows how SIRT6 stops sitting on the L1 promoter, allowing L1 expression to occur.
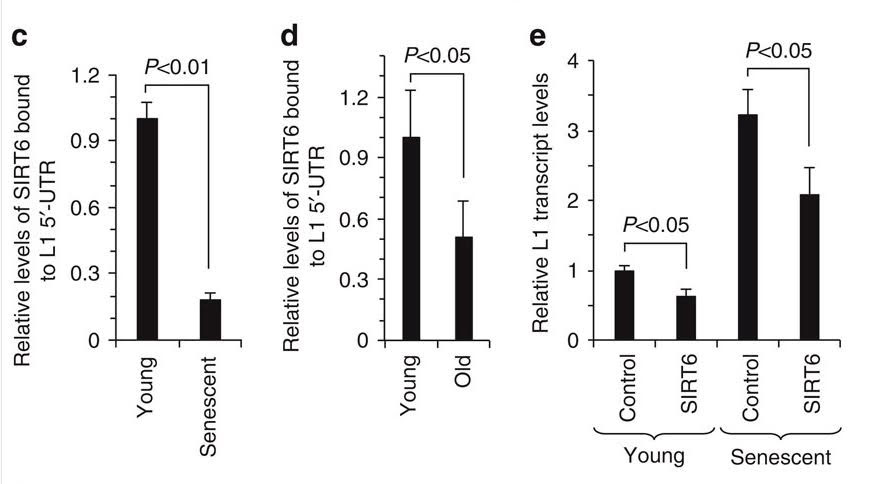
References: 2014 SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age
7. Inflammation triggers transposable element expression
– diseases with inflammation show high levels of expression of L1 ORF2 and L1ORF2 transcripts. Models of Skin inflammation can also be created by triggering DNA transposon-directed inflammatory pathways.
There is strong evidence that chronic inflammation triggers TE transcription in both plants and animals, along with heat shock, oxidative and other stresses. In humans, this has been documented in several diseases, including rheumatoid arthritis. This is all consistent with points made earlier about stresses and TE expression
Here is an illustration from Capy, et al, in Nature, 2000 Stress and transposable elements: co-evolution or useful parasites?
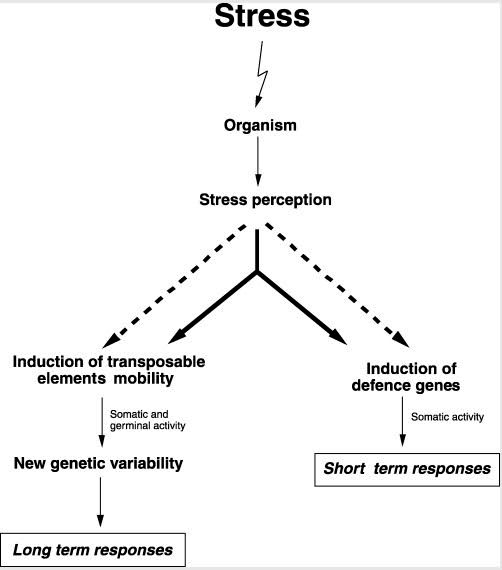
References:
2000 “Stress and transposable elements: co-evolution or useful parasites?
1997 The expression of the tobacco Tnt1 retrotransposon is linked to plant defense responses
1992 Induction of the mobile genetic element Dm-412 transpositions in the Drosophila genome by heat shock treatment “Males of a Drosophila melanogaster isogenic line with a mutation of the major gene for radius incompletus (ri) were treated by standard light heat shock (37 degrees C for 90 min) and by heavy heat shock (transfer of males from 37 degrees C for 2 hr to 4 degrees C for 1 hr and back; this procedure was repeated three times). In the F1 generation of treated males mated with nontreated females of the same isogenic line, mass transpositions of copia-like mobile genetic element Dm-412 were found. The altered positions of the element seem nonrandom; five “hot spots” of transposition were found. Probabilities of transpositions were estimated after light heat shock and heavy heat shock and in the control sample. These probabilities were, respectively, 3.4 x 10(-2), 8.7 x 10(-2), and less than 4.1 x 10(-4) transpositions per genome per occupied position per generation. Therefore, as a result of heat shock treatment, the probabilities of transpositions were two orders of magnitude greater than those of the control sample in the next generation after induction. Comparison of the results with those after stepwise temperature treatment shows that the induction depends on the intensity of the stress action (temperature treatment) rather than on the type of the stress action.”
2012 Epigenetic Epidemiology of Inflammation and Rheumatoid Arthritis
2013 Association between blood pressure and DNA methylation of retrotransposons and pro-inflammatory genes Some of the conclusions of this study are of general interest “Results: An increase in inter-quartile range (IQR) in the methylation of Alu elements was associated with an increase of 0.97 mm Hg in diastolic blood pressure (DBP) (95% CI 0.32–1.57), but no such association was observed for long interspersed nuclear element-1 (LINE-1). We also found positive associations between DBP and methylation of the genes for toll-like receptor 2 (TLR2) and inducible nitric oxide synthase (iNOS), and a negative association between DBP and methylation of the gene for interferon-γ (IFN-γ). Associations between methylation and systolic blood pressure (SBP) were weaker than those between methylation and DBP. Bayesian mixed-effects structural equation model results were similar for both DBP and SBP models. — Conclusions: The results of our study suggest that changes in DNA methylation of some pro-inflammatory genes and retrotransposable elements are related to small changes in BP.” It is unclear whether the association represents causality in either direction.
2008 Epigenetic Regulation of Inflammatory Response and Relevance eo Common Diseses
1994 Genome and stresses: Reactions against aggressions, behavior of transposable elements
8. Germ lines and stem cells are protected against TE expression – role of the Piwi proteins and piRNA pathways
Since TEs can wreak so much genetic havoc, evolution has had to provide mechanisms to keep germline cells so that the germline can remain relatively constant from generation to generation. Likewise, stem cells that may be needed to replace somatic cells require protection. It appears that in a number of species silencing TEs in such cells is in part or in whole accomplished by expression of Piwi proteins.
The 2011 publication Uniting germline and stem cells: the function of Piwi proteins and the piRNA pathway in diverse organisms relates “The topipotency of the germline is the full manifestation of the pluri- and multipotency of embryonic and adult stem cells, thus the germline and stem cells must share common mechanisms that guarantee their multipotentials in development. One of the few such known shared mechanisms is represented by Piwi proteins, which constitute one of the two subfamilies of the Argonaute protein family. Piwi proteins bind to Piwi-interacting RNAs (piRNAs) that are generally 26 to 31 nucleotides in length. Both Piwi proteins and piRNAs are most abundantly expressed in the germline. Moreover, Piwi proteins are expressed broadly in certain types of somatic stem/progenitor cells and other somatic cells across animal phylogeny. Recent studies indicate that the Piwi-piRNA pathway mediates epigenetic programming and posttranscriptional regulation, which may be responsible for its function in germline specification, gametogenesis, stem cell maintenance, transposon silencing, and genome integrity in diverse organisms.”
The 2014 publication 2014 Homeland security in the C. elegans germ line: insights into the biogenesis and function of piRNAs relates: “While most eukaryotic genomes contain transposable elements that can provide select evolutionary advantages to a given organism, failure to tightly control the mobility of such transposable elements can result in compromised genomic integrity of both parental and subsequent generations. Together with the Piwi subfamily of Argonaute proteins, small, non-coding Piwi-interacting RNAs (piRNAs) primarily function in the germ line to defend the genome against the potentially deleterious effects that can be caused by transposition. Here, we describe recent discoveries concerning the biogenesis and function of piRNAs in the nematode Caenorhabditis elegans, illuminating how the faithful production of these mature species can impart a robust defense mechanism for the germ line to counteract problems caused by foreign genetic elements across successive generations by contributing to the epigenetic memory of non-self vs. self.”
The piRNA pathway is active in us as well as in c-elegans and fruit flies. The 2014 publication Piwi is required in multiple cell types to control germline stem cell lineage development in the Drosophila ovary reports “The piRNA pathway plays an important role in maintaining genome stability in the germ line by silencing transposable elements (TEs) from fly to mammals. As a highly conserved piRNA pathway component, Piwi is widely expressed in both germ cells and somatic cells in the Drosophila ovary and is required for piRNA production in both cell types. In addition to its known role in somatic cap cells to maintain germline stem cells (GSCs), this study has demonstrated that Piwi has novel functions in somatic cells and germ cells of the Drosophila ovary to promote germ cell differentiation. — This study has revealed new germline requirement of Piwi in controlling GSC maintenance and lineage differentiation as well as its new somatic function in promoting germ cell differentiation. Therefore, Piwi is required in multiple cell types to control GSC lineage development in the Drosophila.
Piwi seems to work via surveillance of the genome for extraneous DNA, epigenetically silencing it when it sees it.
Addittional references
2014 Silent no more: Endogenous small RNA pathways promote gene expression. “Endogenous small RNA pathways related to RNA interference (RNAi) play a well-documented role in protecting host genomes from the invasion of foreign nucleic acids. In C. elegans, the PIWI type Argonaute, PRG-1, through an association with 21U-RNAs, mediates a genome surveillance process by constantly scanning the genome for potentially deleterious invading elements. Upon recognition of foreign nucleic acids, PRG-1 initiates a cascade of cytoplasmic and nuclear events that results in heritable epigenetic silencing of these transcripts and their coding genomic loci. If the PRG-1/21U-RNA genome surveillance pathway has the capacity to target most of the C. elegans transcriptome, what mechanisms exist to protect endogenous transcripts from being silenced by this pathway? In this commentary, we discuss three recent publications that implicate the CSR-1 small RNA pathway in the heritable activation of germline transcripts, propose a model as to why not all epialleles behave similarly, and touch on the practical implications of these findings.”
2012 Function, targets and evolution of Caenorhabditis elegans piRNAs
2015 piRNAs and piRNA-Dependent siRNAs Protect Conserved and Essential C. elegans Genes from Misrouting into the RNAi Pathway “piRNAs silence foreign genes, such as transposons, to preserve genome integrity, but they also target endogenous mRNAs by mechanisms that are poorly understood. Caenorhabditis elegans piRNAs interact with both transposon and nontransposon mRNAs to initiate sustained silencing via the RNAi pathway. To assess the dysregulation of gene silencing caused by lack of piRNAs, we restored RNA silencing in RNAi-defective animals in the presence or absence of piRNAs. In the absence of piRNAs and a cellular memory of piRNA activity, essential and conserved genes are misrouted into the RNAi pathway to produce siRNAs that bind the nuclear Argonaute HRDE-1, resulting in dramatic defects in germ cell proliferation and function such that the animals are sterile. Inactivation of RNAi suppresses sterility, indicating that aberrant siRNAs produced in the absence of piRNAs target essential genes for silencing. Thus, by reanimating RNAi, we uncovered a role for piRNAs in protecting essential genes from RNA silencing.”
2013 Biology and Mechanisms of Short RNAs in Caenorhabditis elegans
2012 Extremely stable Piwi-induced gene silencing in Caenorhabditis elegans “In recent years, the Piwi pathway has been shown to regulate the silencing of mobile genetic elements. However, we know little about how Piwi pathways impose silencing and even less about trans-generational stability of Piwi-induced silencing. We demonstrate that the Caenorhabditis elegans Piwi protein PRG-1 can initiate an extremely stable form of gene silencing on a transgenic, single-copy target. This type of silencing is faithfully maintained over tens of generations in the absence of a functional Piwi pathway. Interestingly, RNAi can also trigger permanent gene silencing of a single-copy transgene and the phenomenon will be collectively referred to as RNA-induced epigenetic silencing (RNAe). RNAe can act in trans and is dependent on endogenous RNAi factors. The involvement of factors known to act in nuclear RNAi and the fact that RNAe is accompanied by repressive chromatin marks indicate that RNAe includes a transcriptional silencing component. Our results demonstrate that, at least in C. elegans, the Piwi pathway can impose a state of gene silencing that borders on ‘permanently silent’. Such a property may be more widely conserved among Piwi pathways in different animals.”
9. Knocking out the Insulin/IGF pathway in C. elegans silences transposons by RNAi,
Piwi-piRNA pathway, and by TDP-1
As observed above, nematodes contain transposable elements in their genome. Like humans, they have the Piwi-piRNA pathway which silences transposons. Defects in the Piwi/piRNA pathway lead to the re-expression of transposons and immediate sterility. Although there are many reasons that have been proposed for why genetic blockade of the Insulin/IGF-1 pathway increase life span in all organisms studied to date, the main explanation that has been given for the benefit of this intervention has been the nuclear localization of the FOXO3a transcription factor (in humans) or the nuclear localization of DAF-16 in C. elegans (the nematode version of FOXO3a). The targets of FOXO3a transcription factor include apoptosis genes and genes that promote cellular resistance to oxidative stress and other cell stressors.
In addition, knocking out the Insulin/IGF-1 pathway promotes retrotransposon silencing via two RNA-based methods.
· The first RNA-based method of slicing transposons found to be activated by fasting or Insulin/IGF-1 genetic knockout was the Piwi-piRNA pathway. This was controlled by the gene prg-1.
· However a recent paper from UNC, Chapel Hill showed that reduced insulin/IGF-1 signaling promoted germ cell immortality via a 2nd pathway, involving RNAI methods. In these studies, they “knocked out” prg-1 gene in C. elegans, and now these prg-1 mutants could not “turn on” the Piwi/piRNA pathway. However, these cells were still able to repress transposon expression by the RNAi pathway. Here is a diagram of how this works:

Illustration reference: 2014 Reduced Insulin/IGF-1 Signaling Restores Germ Cell Immortality toCaenorhabditis elegans Piwi Mutants
This study showed that there was more than one way of silencing transposons in C. elegans and demonstrated the health benefits of fasting. They found that brief periods of starvation in C. elegans extended the transgenerational lifespan of prg-1 mutants by stimulating the DAF-16 (FoxO) transcription factor. Constitutive activation of DAF-16 by fasting immortalized germ line cells by silencing repetitive elements (repeats and transposons) via RNAi proteins in these prg-1 mutants (i.e. the RNAi pathway that is parallel to the Piwi-piRNA pathway) as well as by histone H3K4 demethylase activation.
Again, reference: 2014 Reduced Insulin/IGF-1 Signaling Restores Germ Cell Immortality toCaenorhabditis elegans Piwi Mutants
So, down-regulating the Insulin/IGF-1 pathway may have a completely independent way of dealing with transposable elements that has nothing to do with RNAi or the Piwi-piRNA pathway. This involves TDP-1 (TDP-43 in humans), a pathway that is “broken” in patients with familial ALS. DAF-16 (FoxO) up regulates the tdp-1 gene in C. elegans. TDP-1 is a DNA binding protein implicated in several neurodegenerative diseases, but exactly how it works is not clear. What is clear is that it is upregulated by heat stress and DAF-16 (i.e. down regulating the Insulin/IGF-1 pathway).
Here are some additional references on this:
2012 TDP-1/TDP-43 Regulates Stress Signaling and Age-Dependent Proteotoxicity in Caenorhabditis elegans
In another study, researchers found that the C. elegans ortholog to the human TDP-43 was also involved with preventing the accumulation of double stranded RNA in C. elegans. TDP-43 is a nuclear protein in human cells, but “gets stuck” in the cytoplasm of patients with familial ALS and FTD/ALS spectrum disorders. As a consequence they develop frontotemporal dementia and amyotrophic lateral sclerossis. In C. elegans, TDP-1 is also found in the nucleus and it prevents dsRNA from accumulating, reduces the frequency of A-to-I editing, and promotes the degradation of dsRNA. Here is an illustration of this in human cells (TDP-43 function):

Image source: 2014 TDP‐1, the Caenorhabditis elegans ortholog of TDP‐43, limits the accumulation of double‐stranded RNA
More references:
2012 TDP-1/TDP-43 Regulates Stress Signaling and Age-Dependent Proteotoxicity in Caenorhabditis elegans
2014 RNA-Processing Protein TDP-43 Regulates FOXO-Dependent Protein Quality Control in Stress Response
Conclusion: The evidence is overwhelming – down regulating the Insulin/IGF-1 pathway suppresses transposable element expression and increases the degradation of double-stranded RNA molecules from endogenous retroviral expression (ERV) and from exogenous retroviruses. There are two of these pathways that are dependent on small RNAs – the RNAi pathway and the Piwi-piRNA pathway. Fasting and caloric restriction work via both of these. In addition, the TDP-1 DNA binding protein also works to limit the accumulation of double stranded RNA molecules within cells. In humans, the TDP-1 ortholog is TDP-43, which is implicated in familial FTD/ALS. Thus targeting the insulin/IGF-1 pathway in humans may help with preventing ALS and other similar diseases like Frontotemporal Dementia by reducing transposable element expression.
10. Transposable element endonuclease (ORF2) induces double stranded DNA breaks (DSBs) via endonuclease “nicking” of DNA – This is a mechanism of DNA damage independent from the oxidative stress-based free radical theory of aging. TE endonucleases do not require ROS to damage DNA and produce DSBs. This may in part explain why anti-oxidant drugs and supplements failed to “cure” or slow down aging.
Whereas most of the discussion up to this point has centered around insertional mutagenesis of transposable elements, there is a completely different mechanism by which TEs can cause aging and cancer. In the 2nd open reading frame (ORF2) of LINE-1, there is an endonuclease domain (EN) that is responsible for breaking genomic DNA. This enzyme is necessary for re-integrating the L1 transcript after it undergoes reverse transcriptase processing. You can think of the endonuclease as being like an ice pick. Unfortunately, every re-integration attempt by the L1 endonuclease produces multiple “nicks” in the DNA. Some of these “nicks” are single stranded DNA breaks (SSBs) but some are double stranded DNA breaks (DSBs).
This of course triggers the DNA damage response and harnesses the SSB repair pathways or the DSB repair pathways, which includes the hyper activation of PARPs, which use up prodigious amounts of SIRT1, NAD and ATP. Thus a “vicious cycle” of “nicks, SSBs, and DSBs” occurs when LINE-1 ORF2 endonuclease is produced.
Because of this “inefficient nicking” system of the ORF2 endonuclease, some experts have estimated that there are 10-100 times more frequent DSBs cause by L1 activity than there are de novo L1 integration events (Gasior, et al, 2006 The Human LINE-1 Retrotransposon Creates DNA Double-strand Breaks). Specifically, they show evidence that L1 activity may be responsible for 0.4 to 7 double stranded DNA breaks per cell. If so, this implicates L1 activity as a major cause of aging, not due to insertional mutagenesis but due to L1 activity-induced DSBs.
Note that this mechanism of induced DSBs does not require any increase in reactive oxygen species (ROS). This may explain why Denham Harmon’s laboratory experiments failed to show that anti-oxidant administration could slow down aging in normal aged rodents and may explain why anti-oxidant administration in humans does not reduce cancer risk. In other words, ROS may not be the major reason for DSBs…..it may be L1 activity.
References:
2006 The Human LINE-1 Retrotransposon Creates DNA Double-strand Breaks
2010 Somatic expression of LINE-1 elements in human tissues
2014 Potential for genomic instability associated with retrotranspositionally-incompetent L1 loci
1996 Human L1 Retrotransposon Encodes a Conserved Endonuclease Required for Retrotransposition
Here is an illustration which shows how ORF2 produces the endonuclease enzyme which “nicks” DNA, producing a DSB:
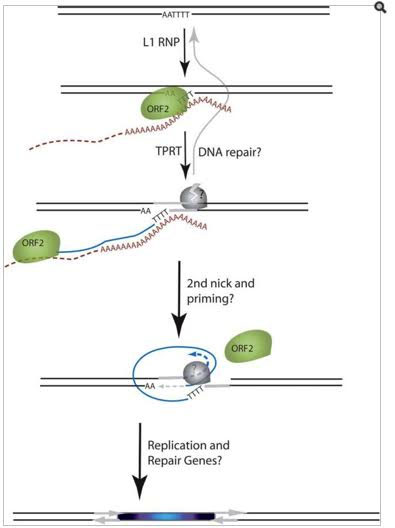
Image and legend source: 2006 The Human LINE-1 Retrotransposon Creates DNA Double-strand Breaks
“Schematic of the L1 TPRT reaction. The top drawing shows a genomic site with a typical L1 endonuclease cleavage site (5’TTTTAA) that is exposed in the nucleus to the L1 RNP, where the ORF2 cleaves the DNA at the A+T-rich consensus sequence. In the second drawing, the T-rich region primes reverse transcription on the L1 mRNA poly(A) tail. The third drawing highlights the reverse transcriptase activity (formation of the blue cDNA) of the ORF2 and the need for a nick to occur on the second strand, made by an unknown source. The segment between the two nicks is highlighted in gray to illustrate the eventual formation of flanking direct repeats by the duplication of these segments. The fourth panel illustrates the possibility that the second-strand synthesis is primed by microhomology-mediated priming, which results in synthesis of a second copy of the gray segment (dotted arrow). Replication from the gray arrow completes synthesis across the 2nd strand of the cDNA creating a new L1 insert and completing synthesis across the other side of the direct repeat.”
Here is another illustration of how both transcription of the functional L1 locus (FL1mRNA) or the splice variant that only contains ORF2 can cause DNA mutations:
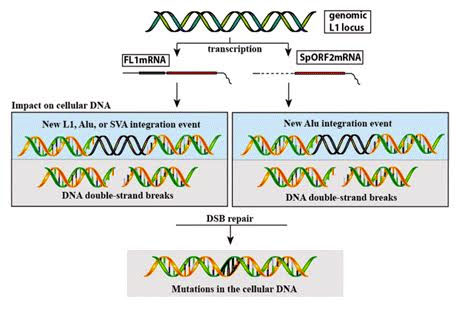
Image source: 2015 Somatic expression of LINE-1 elements in human tissues
Here is an illustration that shows why you need a competent L1 to create endonuclease from ORF2. It also summarizes the three deleterious mechanisms by which L1 retrotransposons can create DNA havoc, cancer and aging.
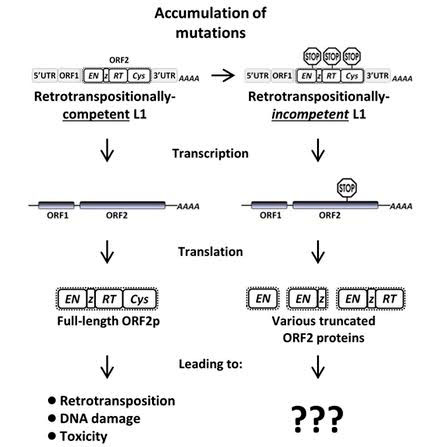
Image source: 2014 Potential for genomic instability associated with retrotranspositionally-incompetent L1 loci
11. Cancer is manifested by dramatic increases in transposable element expression
There is very strong evidence that cancer cells express transposable elements and that cancer cells that have progressed to late stages of chemotherapy and radiation resistance have very high levels of TE expression. The exact role that TE expression plays is still a mystery, however. Part of this mystery may have been recently solved by some researchers who found double-stranded hybrid nucleic acid strands (DNA:RNA DS hybrids) that contain both LINE-1 and Alu elements. When cancer cells are treated with LINE-1 reverse transcriptase inhibitor drugs, such as efavirenz (EFV), five amazing effects occur simultaneously in the RT-inhibitor treated cancer cells:
– the DNA:RNA double-stranded hybrid molecules containing LINE-1 and Alu copies fail to form
– cancer cell proliferation is repressed
– coding genes that were silenced are re-expressed
– many microRNAs that were dysregulated are re-expressed
– ultra conserved regions (UCRs) are re-expressed
What is even more fascinating is that the RT inhibitor-sensitive microRNAs and UCRs that are re-expressed are significantly associated with Alu elements. This was just reported in 2013 and opens up an exciting new field in cancer research (Sciamanna, et.al, 2013 A tumor-promoting mechanism mediated by retrotransposon-encoded reverse transcriptase is active in human transformed cell lines).
Several authors have suggested that these same RT inhibitor drugs may also be used to treat aging, since the expression of LINE-1 elements dramatically increases with aging in S. cerevisiae (Maxwell, et al, 2011), in the Drosophila brain (Li, 2013), in the mouse liver (Cecco, et al, 2013), in the mouse skeletal muscle (Cecco, et.al, 2013), and in aging human cells (Belancia, et al, 2010). To date, no animal experiments or human clinical trials have shown that RT inhibitor drugs prolong life span, however.
Here is an illustration for how the RISC complex represses TE expression in normal cells, in early tumors, and chemotherapy resistant cancers.
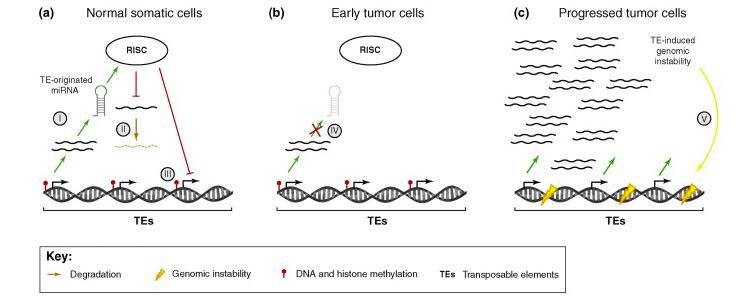
Image source: 2010 Repression of transposable-elements – a microRNA anti-cancer defense mechanism?
12. LTR transposable elements contain G-quadruplex sequences at specific distances from their endogenous TE promoter that may regulate LTR-mediated gene expression –G-quadruplexes in the 3′-UTRs and 5′-UTRs of LINE-1 elements may affect expression of neighboring genes
G-quadruplexes are common, but complex tertiary “3D” structures found in G-rich sequences of DNA that form stable structures using a potassium cation in their center and a series of hydrogen bonds called “Hoogsteen bonds”. G-quadruplexes are formed within the 3′ overhang region of the telomeric DNA and help stabilize/protect the tip of the telomere. TERRA, the long non-coding RNA that associates with the tellomere to transcriptionally silence telomerase, also has a G-quadruplex in it. For more on this fascinating topic, read the blog that Dr. Vince Giuliano and I recently posted on this website: 2015 G-quadraplexes.
There are hundreds of thousands of potential G-quadruplex structures within our DNA sequences and they are evolutionarily conserved. One of the many reasons why the BRCA1 gene (we call it the “Angelina Jolie Gene” now) is so important in preventing breast cancer is because it controls G-quadruplex formation in the telomere (via its control over Rad50). Perhaps the most shocking thing about G-quadruplexes, however, is that 90% of the expression of the c-Myc oncogene is controlled by a G-quadruplex structure just upstream from its promoter. c-Myc is the most important oncogene that gets up-regulated in cancer. Thus it may be that this upstream G-quadruplex may be the reason for the hereto unexplained up-regulation of c-Myc that occurs in the majority of tumors.
A recent study showed that there probably are G-quadruplex structures within transposable elements (see references below). Quadruplex DNA can affect the whole genome as it represents a barrier for DNA replication. This is why helicases like Werner’s Rec-helicase is so important in the proper functioning of DNA. When the Werner helicase gene is mutated, accelerated aging occurs and the phenotype of the progeria Werner’s syndrome almost exactly “mimics” normal aging, but at an accelerated rate (the children with the progeria die at an early age).
The link between TEs and G-quadruplexes is via the protein ATRX. ATRX is part of a family of SWI/SNF proteins that are chromatin modulators. ATRX interacts with Heterochromatin Protein 1 (HP1) the protein that interacts with nuclear corepressor protein 1 (KAP1), which is the target of SIRT6-mediated transposable element silencing. ATRX and HP1 also interact with trimethylated lysine 4 and lysine 9 in histone 3 (i.e. H3K4me3 and H3k9me3). H3K9me3 plays a role in transposon silencing.
References
2013 Quadruplex-forming sequences occupy discrete regions inside plant LTR retrotransposons
Here is an illustration of how LTRs work in plant LTR retrotransposons and in human LINE-1 elements:
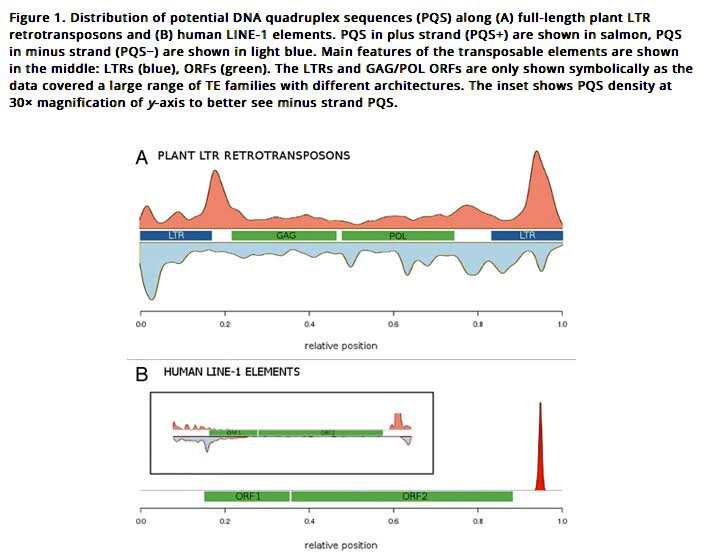
Image source: 2014 Quadruplex-forming DNA sequences spread by retrotransposons may serve as genome regulators
Here is an illustration of how G-quadruplexes could play a role in blocking transposable element retrotransposition, inhibiting ectopic recombination, and forming truncated 5′ copies that would not have made functional copies.
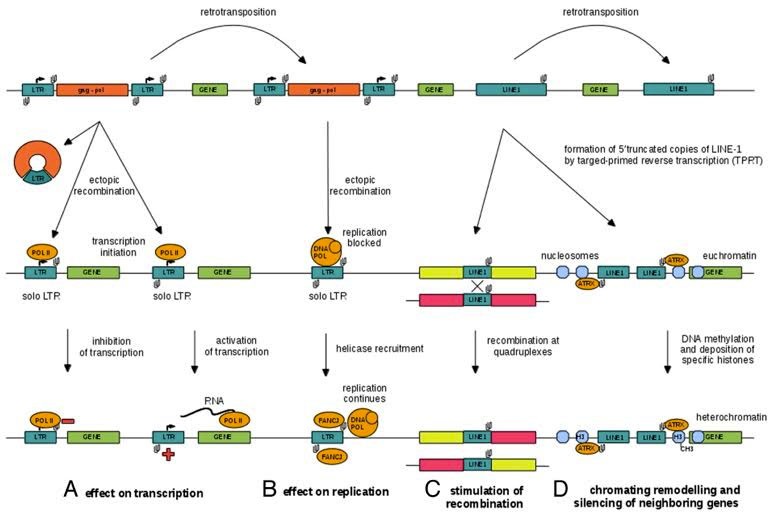
Image and text source: 2014 Quadruplex-forming DNA sequences spread by retrotransposons may serve as genome regulators ” Figure 2. A model of DNA quadruplex spreading by retrotransposition and the assumed biological functions of DNA quadruplexes in gene regulation and genome organization: (A) negative and positive effect on transcription via interfering with RNA extension or separating DNA strands, respectively, (B) inhibitory effect on DNA replication that can be overcome by binding of FANCJ-type helicases, (C) serving as hotspots for recombination and (D) the role in chromatin remodeling by attracting ATRX-like proteins that are associated with heterochromatin or heterochromatin boundaries.”
13. Transient LINE-1 over-expression induces cell senescence or apoptosis in normal healthy cells – LINE-1 expression is “toxic” to cells
There is recent evidence that merely expressing L1 can induce cellular senescence or apoptosis in normal cells. Belancio and colleagues in 2010 showed that expression of the wild type L1 element (mRNA) or the splice variant (SpORF2) in normal human fibroblasts and adult stem cells leads to a sensecent-like phenotype. Even low levels of L1 activity have the potential to contribute to genetic instability, aging, and age-related diseases like cancer. When they looked at old normal cells from the esophagus, prostate, stomach, and heart muscle, they found levels of L1 expression that were 80% (esophagus), 50% (prostate), 150% (stomach), and 200% (heart muscle) of the levels of L1 expression in HeLa cells. Thus it is clear that somatic cells express L1. In the testes, there is a dramatic increase in L1 expression that is an order of magnitude above HeLa cells, as well as old normal somatic cells. Several authors have shown that germ cell tissues (testes) display a 10-fold increase in transcriptional activation of the L1 promoter by the testes-determining factor gene, SRY. Even mesenchymal stem cells express L1 transcripts, but at low levels that are 20% of what is seen in MCF7 cancer cells.
References:
2010 Somatic expression of LINE-1 elements in human tissues “Tests of the senescence-associated β-galactosidase expression suggest that expression of exogenous full-length L1, or the SpORF2 mRNA alone in human fibroblasts and adult stem cells triggers a senescence-like phenotype, which is one of the reported responses to DNA damage. In contrast to previous assumptions that L1 expression is germ line specific, the increased spectrum of tissues exposed to L1-associated damage suggests a role for L1 as an endogenous mutagen in somatic tissues. These findings have potential consequences for the whole organism in the form of cancer and mammalian aging.”
2006 Accumulation of Senescent Cells in Mitotic Tissue of Aging Primates
14. Melatonin suppresses LINE-1 expression, as does sleep and turning off the lights at night – Light exposure at night and disruption of circadian rhythms prevents the circadian-based silencing of L1 retrotransposons
For a long time, it has been well-known that the expression of LINE-1 element mRNAs is increased in cancer. It has also been known for quite some time that night shift workers have and increase risk of cancer and female airline flight attendants who are constantly disrupting circadian rhythms have a 1.4 o 1.6 fold higher risk of breast cancer. However, the exact mechanism by which working at night and disrupting circadian rhythms induces cancer remains a mystery. Well, the mystery may be solved by LINE-1.
A fascinating paper recently showed that light exposure at night is associated with an inhibition of mobilization of L1 in cultured cells through the down regulation of L1 mRNA and ORF1 protein. Specifically, the mechanism of action was that activating the melatonin receptor 1 inhibited the mobilization of L1. They also showed that the administration of exogenous melatonin inhibited L1 mobilization in cultured cells. Their conclusion was that the decreased melatonin production in shift workers increased L1 transcription into mRNA and increased L1 mobilization. This is one of the first papers to link sleep, transposable elements, and cancer. However, many papers prior to this have linked melatonin to a reduced risk of breast cancer via a MT-1 receptor-mediated mechanism. In these papers, other molecular mechanisms of action besides LINE-1 repression have been shown to exist. This includes the regulation of the DNA repair mechanism by melatonin called the “Nucletoide Excision Repair” (NER) pathway, which repairs single stranded DNA breaks.
The good news is that exogenous melatonin supplementation is well-absorbed, inexpensive, and has very few side effects besides being sleepy. Not only does it suppress LINE-1 transcription, it also preserves SIRT1 expression in the hippocampus, even in sleep deprived mice. It is likely that melatonin works via both SIRT1 and SIRT6 to suppress LINE-1 expression.
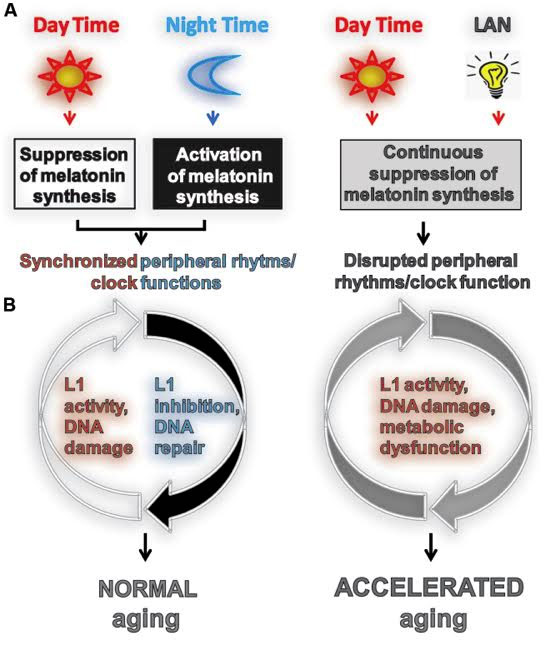
Image source “FIGURE 1. Light exposure at night accelerates aging by impeding or enhancing processes associated with aging. (A) Usually aging involves normal light exposure that is characterized by alternating intervals of light and dark over a 24-h period, which result in circadian production of nocturnal melatonin. This leads to the synchronization of peripheral clock (PC) function controlling many biochemical processes in cells including L1 expression and activity (Deharo et al., 2014) and the DNA damage response (DDR). (B) Exposure to light at night (LAN) is reported to accelerate aging. LAN blocks nocturnal melatonin production which prevents synchronization of PCs, leading to the disruption of timely function of many biochemical processes in cells including L1 expression and activity, DDR, and metabolism.”
References:
2015 The aging clock and circadian control of metabolism and genome stability “It is widely accepted that aging is characterized by a gradual decline in the efficiency and accuracy of biological processes, leading to deterioration of physiological functions and development of age-associated diseases. Age-dependent accumulation of genomic instability and development of metabolic syndrome are well-recognized components of the aging phenotype, both of which have been extensively studied. Existing findings strongly support the view that the integrity of the cellular genome and metabolic function can be influenced by light at night (LAN) and associated suppression of circadian melatonin production. While LAN is reported to accelerate aging by promoting age-associated carcinogenesis in several animal models, the specific molecular mechanism(s) of its action are not fully understood. Here, we review literature supporting a connection between LAN-induced central circadian disruption of peripheral circadian rhythms and clock function, LINE-1 retrotransposon-associated genomic instability, metabolic deregulation, and aging. We propose that aging is a progressive decline in the stability, continuity, and synchronization of multi-frequency oscillations in biological processes to a temporally disorganized state. By extension, healthy aging is the ability to maintain the most consistent, stable, and entrainable rhythmicity and coordination of these oscillations, at the molecular, cellular, and systemic levels.”
2015 Melatonin: an inhibitor of breast cancer
15. SIRT6 – An important silencer of transposable element expression and a candidate “longevity gene“
SIRT6 has a unique role in silencing transposable elements. Specifically, it silences transposable elements, telomeric DNA, and satellite DNA by ribosylating KAP1, a nuclear co-repressor protein that organizes and compacts chromatin into heterochromatin. It is not surprising then that in GWAS studies, SNPs in the SIRT6 gene have showed up as being “longevity SNPs.” Recently, researchers from the University of Iowa looked at a elderly cohort aged > 65 for variations in the Sirtuin genes (SIRT1-7), FOXO3a, and SOD3. Of these genes, by far the most significant SNPs were found in the SIRT6 genes. “Good SNPs” in the SIRT6 gene conferrred a longevity advantage of over 5 years. No other gene SNPs gave a longevity advantage of more than 1.6 years.
Specifically rs107251 showed that individuals with the homozygous “CC” or heterozygous “CT” genotype lived 5.5 and 5.9 years longer than those with the homozygous “TT” genotype for this SIRT6 SNP.
References:
2014 SIRT6 Minor Allele Genotype Is Associated with >5-Year Decrease in Lifespan in an Aged Cohort
16. Longevity is associated with gene polymorphisms that may play a role in reducing TE activity and reducing inflammation – IFN-gamma SNPs IL-6 SNPs, ADARB1 and ADARB2 SNPs, YTHDF2 SNPs, and SIRT6 SNPs may all work to extend lifespans via transposable elements
IFN-gamma and IL-6 SNPs
Inflammation triggers transposable element expression and this may be a major link between inflammation and aging. Only SNPs in genes that trigger transcription factors that can activate LTRs (IFN-gamma and IL-6) are associated with longevity. For instance, a SNP in the IFN-gamma gene that reduces IFN-g function is associated with Longevity and a SNP in IL-6 gene that reduces IL-6 mediated signaling is associated with Longevity. It has been known for a long time that inflammation is associated with accelerated aging. This has often been referred to as “inflammaging”. There is now strong evidence that Transposable elements become activated and TE transcription increases with inflammation and cellular stress.
When studies showed that SNPs in IFN-gamma reduced inflammation, this was not surprising. If TE expression shortens life span, this could be one of the mechanisms by which the molecular mechanism by which inflammation, aging, and shortened lifespan were linked. Interferons are known to be produced in response to viral infections and trigger a host of pathways that help the cell survive the viral invasion.
Unfortunately, some of the inflammatory transcription factors produced by IFN-gamma can activate LTRs found near human genes. These inflammatory transcription factors can use the LTR as a “landing strip” and activate nearby genes that can produce cancer and aging. This is why chronic viral infections are associated with an increased risk of cancer and accelerated aging. For instance, chronic CMV infections increase IFN-gamma secretion and this results in accelerated telomere shortening.
In a study of Italian centenarians, polymorphisms in the first intron of the interferon-gamma (IFN-g) gene that decreased IFN-gamma production were associated with longevity (the +874A allele), whereas the “T” copy of the +874T SNP which was associated with increased IFN-gamma production was found less frequently in centenarians. Since then, other researchers have elucidated the mechanisms of action for the +874A polymorphism’s “longevity effect” – it appears to be associated with a micro satellite repeat in this intron. When this micro satellite repeat was expanded, it resulted in a down regulation in the IFN-gamma gene expression. The general theory for how IFN-gamma could reduce longevity was hypothesized to be mediated by increased inflammation, according to the authors of this paper. If the “longevity allele” in the IFN-gamma SNP decreased IFN-gamma production by expanding the micro satellite repeat, this could be a plausible mechanism of action. However the effect of IFN-gamma on activating transcription factors associated with LTRs is entirely possible with this mechanism of action. In other words, +874A allele reduced inflammation. Researchers in Belfast, UK also found polymorphisms in a micro satellite repeat in intron 1 of the IFN-gamma gene to be enriched in centenarians in the UK. This same group also found polymorphisms in the IL-6 gene that were associated with longevity. Specifically, the -174CC allele of the IL-6 gene was enriched in centenarians, whereas the -174GG allele were not enriched in centenarians. Again, there is no question that “inflammaging” is one of the hallmarks of aging.
SNPS in several other inflammatory cytokines were also analyzed, but no correlation with longevity was found for these other SNPs (IL-2, IL-8, IL-10, and IL-12). A group from Finland also found no association between longevity and polymorphisms in IL-10, but they also found no association of SNPs in IL-6 or TNF-alpha and lifespan. Obviously more studies in this arena need to be done before any conclusions are reached.
References:
2009 The role of transposable elements in the regulation of IFN-λ1 gene expression
2003 Study_of_age-association_with_cytokine_gene_polymorphisms_in_an_aged_Irish_population problem with link
17. The human brain is a very active site of L1 transcription and insertions into neurons, in a cell-specific manner that produces gene mosaicism – Researchers have already shown that L1 retrotranspositions not only copy their own genomes — they also copy human genes such as AKT3 and APP. This produces a “gene dosage problem” which can account for hemimegalencephly, increases in Amyloid-beta production, and other diseases.
Recent studies using Next Generation Sequencing (NGS) conduced on a single cell analysis has showed that somatic neurons (i.e. post mitotic cells) undergo L1 retrotransposition at a rate of 0.04 to 0.07 inserts per neuron (Evrony, et al, 2012 Single-Neuron Sequencing Analysis of L1 Retrotransposition and Somatic Mutation in the Human Brain). With an estimated 50 trillion cells in the human body, it is estimated that 4 billion neurons per brain may contain somatic L1 insertions. Not only can L1 “copy machines” copy their L1 genomes — they can also copy adjacent human genes. The consequences for human aging could be most serious.
Reference: 2014 The aging clock and circadian control of metabolism and genome stability
18. TE-originated genetic mosaicism – a likely cause of Alzheimer’s disease
The beta amyloid overload so characteristically seen in Alzheimer’s disease may be largely due to TE-originated genetic mosaicism which leads some cortical neurons to have large copy numbers of the amyloid precursor protein (APP) gene.
Another major breakthrough in the understanding of the link between disease and transposable elements was published this year by a group from Scripps Institute in La Jolla, California. In February, 2015, Dr. Jerold Chun and colleagues published their data showing that extra copies of the gene that encode for the precursor protein for amyloid beta (APP) was copied when retrotransposons were copied off chromosome 21. The copy number for the APP gene should normally be 2, but in the cerebral cortical neurons harvested from autopsies done on patients who had Alzheimer’s disease, some of the cortical neurons showed a “gene copy amplification” of the APP gene with some cells having as many as 12 copies of APP. The pattern of “genetic mosaicism” that was seen has been described in the brain from earlier work, but this was the first evidence linking over-expression of APP to retrotransposon activity. This may be a major component of the cause or Alzheimer’s disease.
The 2015 publication Genomic mosaicism with increased amyloid precursor protein (APP) gene copy number in single neurons from sporadic Alzheimer’s disease brains reports: ” Previous reports have shown that individual neurons of the brain can display somatic genomic mosaicism of unknown function. In this study, we report altered genomic mosaicism in single, sporadic Alzheimer’s disease (AD) neurons characterized by increases in DNA content and amyloid precursor protein (APP) gene copy number. AD cortical nuclei displayed large variability with average DNA content increases of 8% over non-diseased controls that were unrelated to trisomy 21. Two independent single-cell copy number analyses identified amplifications at the APP locus. The use of single-cell qPCR identified up to 12 copies of APP in sampled neurons. Peptide nucleic acid (PNA) probes targeting APP, combined with super-resolution microscopy detected primarily single fluorescent signals of variable intensity that paralleled single-cell qPCR analyses. These data identify somatic genomic changes in single neurons, affecting known and unknown loci, which are increased in sporadic AD, and further indicate functionality for genomic mosaicism in the CNS.”
Alas, AD is not the only aging-related disease where retrotransposon activity has been implicated. Others include cancer, rheumatoid arthritis, diabetes type I, schizophrenia, infertility, and Amyotrophic Lateral Sclerosis (ALS).
19. Vaccines with cancer-associated ERVS and HIV infection-associated ERVs appear to be safe and immunogenic – This is potentially a therapeutic way to fight cancer and prevent HIV, but only animal studies have been done so far.
One reason why so few people are interested in transposable elements is that they do not understand that unwanted TE expression is a “solvable” problem that might lead to multiple preventative measures and therapies. As an example, cancer and HIV-infected individuals many times do not produce a robust T cell immune response because of a number of “immune evasion” techniques that cancer cells and HIV-infected cells use to evade the immune response against the cancer or HIV virus. A group of researchers at Pfizer in La Jolla are currently working on a novel way of doing something about this. They vaccinated mice with the Human Endogenous Retrovirus (HERV-K) envelope and gag proteins to induce a T cell response. This produced a robust T cell response. They also tried immunizing mice with LINE-1 open reading frame 2 (ORF2). This produced a robust T cell response as well. No tissue damage was noted with these vaccines.
Reference: 2012 Vaccination with Cancer- and HIV Infection-Associated Endogenous Retrotransposable Elements Is Safe and Immunogenic
20. Reverse Transcriptase inhibitors that suppress transposable element retrotransposition may effectively treat cancer – This is potentially a therapeutic way to fight cancer and prevent HIV, but only animal studies have been done so far. These drugs may also have potential to slow aging
Calorie restriction has been proven to reduce age-related increases in TE expression. In the study above, 40% CR reduced the increase in MSAT expression by 50-60% in livers and muscle, but CR did not reduce it to zero. Likewise CR reduced L1 expression by 25-35% in livers and muscle, but CR did not completely reverse the increase in L1 transposable element expression with aging.
There may be other ways to more effectively reduce satellite and transposable element expression. One of them is to use FDA-approved drugs that block the enzyme reverse transcriptase, which is used by the HIV virus but also is used to copy L1s, Alu elements, SVA elements, and other TEs.
An important workshop was held in Erice, Italy, in October, 2013. A select group of experts in the field of aging research were invited to attend this meeting which was titled “Interventions to Slow Aging in Humans: Are We Ready?” The conference published a great review article in the journal, Aging Cell, in April, 2015. Here is the reference for it:
Reference: 2015 Interventions to Slow Aging in Humans: Are We Ready?
In this article, they talked about biomarkers to test efficacy, biomarkers for pro-aging mechanisms, companion biomarkers, safety biomarkers, dietary interventions (CR, fasting, alternative day fasting, methionine restriction, etc.) and pharmacologic interventions that mimicked CR. In this category, they identified the following class of drugs that targeted various CR pathways as follows:
a. mTOR pathway inhibitors
b. Glycolysis inhibitors
c. hGH/IGF-1 pathway inhibitors
d. Sirtuin activators
e. AMPK pathway activators
f. Inhibitors of inflammatory pathways
g. Modulators of epigenetic pathways
h. beta2-adrenergic receptor signaling
i. NDGA
j. statins and ACE inhibitors
k. Hexosamine pathway and glycobiology inhibitors
l. DNA damage signaling drugs
m. stem cell therapy
n. drugs that suppress retrotransposable elements
If you look at the list of authors on this paper, it is a list of the future “hall of fame” in aging research. None of these guys are amateurs and they all are well aware of the futility of things like anti-oxidants, multivitamins, hormone replacement therapy, etc. I thought it was interesting that they included retrotransposable element drugs in the list of strategies for treating aging. Specifically, they mentioned two drugs that were reverse transcriptase inhibitors (lamivudine and adefovir). When I looked these up, I found a paper that was written in 2008 where researchers from UC San Francisco and Toronto, Canada; had already tried nucleoside analogues that inhibited reverse transcriptase enzymes. These drugs were mostly developed and FDA approved in the 1990s to treat HIV. Although some are still used today, the protease inhibitors have largely taken over the market for treating HIV infections.
These reverse transcriptase inhibitors also inhibit the reverse transcriptase encoded by LINE-1 transposable elements. As a result, these FDA-approved drugs inhibit retrotransposition in a human cell lines. They proved this with a reporter assay for LINE-1 retrotransposition. Here are two references showing how FDA-approved reverse transcriptase inhibitors inhibit LINE-1 retrotransposition:
References:
They found that all of the nucleoside inhibitors reduced LINE-1 retrotransposition by as much as 90%, but the non-nucleoside (HIV-1) reverse transcriptase inhibitors like nevirapine did NOT inhibit LINE-1 retrotransposition. Although the doses required for treating HIV infection with these drugs is associated with a lot of side effects, the lower doses used to treat Hepatitis B infections with these drugs is much lower and do not have as much side effects. To date, I do not believe any animal model has been done to see if the nucleoside analog reverse transcriptase inhibitors have any effect on health span or life span in model organisms.
21. Retrotransposon activity is likely to be a MAJOR cause of aging
This is the last point I will make on this subject. I think that the evidence now is irrefutable. L1 activity causes aging by 3 mechanisms:
1) retrotransposition-induced mutagenesis, silencing of genes, and gene copy number increase,
2) L1 endonuclease-mediated double stranded DNA breaks due to the multiple DNA “nicks” that each copy of ORF2 produces, and
3) direct toxicity of L1 transcripts which can trigger pathways that lead to cellular senescence and apoptosis.
As a factor in aging, the relative impact of TEs is likely to be much larger in older people, because: a) transposon activity is much larger in older cells, b) metabolic factors that inhibit TE activity including NAD+ and SIRT1 are scarcer in older people and are drawn off by the larger number of single and double strand DNA breaks that goes along with aging. This is with negative health and pro-aging consequences for other key processes, such as maintenance of mitochondrial health and mitochondrial biogenesis, and avoidance of Warburg-type metabolism, c) increasing activation of endogenous TEs associated with global genome demythelation that comes with aging, and d) increases in copy numbers of some pro-inflammatory and other pro-aging genes in certain cells associated with increasing genetic moscisism.
HERVs and LTR remnants of HERVs can play a role in aging because of their effects in the inflammatory pathways, where they can turn on inflammation by using the LTRs as “alternative promoters” for NF-kB and many other inflammatory transcription factors.
There may be a role for LTRs in chronic viral infections that induce accelerated aging as well, in HIV, hepatitis, CMV, and EBV chronic infections. Alu elements play a role in aging that is much more complex and may include the role of paired Alu elements producing double stranded motifs that allow ADAR to do A-to-I editing. In addition, Alu motifs can disrupt genes by insertional mutagenesis, gene silencing, and disruption of gene regulatory networks. Then there is the role for the RNA Alu motif on the long noncoding RNA called ANRIL, which plays a central role in the genetic risk of all atherosclerotic diseases, cancer, diabetes, and Alzheimer’s disease.
The combined effects of L1s, HERV LTRs, and Alu elements on aging make transposable elements one of the greatest overlooked components of the pathogenesis of aging. All of this evidence on transposable elements is validated by the fact that caloric restriction, fasting, and genetic knock-out of the Insulin/IGF-1 pathway silences retrotransposon activity via RNAI and PIWI-piRNA pathways, as well as activating the gene for the DNA binding protein called TDP-43. This is “good news”, since caloric restriction and fasting are free, effective methods to silence transposons. In addition, melatonin, SIRT6 activators, biotin, histone deacetylase inhibitors, histone acetyl transferase inhibitors, and Sirtuin activators may all be promising strategies to suppress TE expression. Last of all, vaccines against ORF2 and HERV proteins are being developed to prevent cancer and HIV infections. So far, they show no toxicity. Reverse transcriptase inhibitors look like a promising strategy to deal with TE-mediated aging as well. Here are the best references that support this view:
2015 The mechanism of ageing: Primary role of transposable elements in genome disintegration
2009 The impact of retrotransposons on human genome evolution
2015 Interventions to Slow Aging in Humans: Are We Ready?
2013 Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues
2015 Intrinsic retroviral reactivation in human preimplantation embryos and pluripotent cells
