
By Vince Giuliano
Art KOUKOU by Vince Giuliano

I. INTRODUCTION AND BACKGROUND
YOUNGING processes in the human body are like AGING processes in reverse. I have suggested that there are natural YOUNGING processes normally going on in mature humans, along with AGING processes. What we experience is normal aging is the result of how the YOUNGING and AGING processes balance against each other over time. Normally of course we seem to get older, though we all accept that aging can be greatly accelerated by certain disease and stress conditions.
I have asserted that the scientific basis now exists for creating conditions which can induce YOUNGING processes sufficient to outweigh aging processes in healthy mature and older persons. In other words it is likely that some of us can soon cast ourselves into mostly aging in reverse, on a temporary basis at least. As I have asserted previously, I believe I’m already doing that. The suggestion is radical, but is supported by very real science. I do not think YOUNGING and its implications are yet adequately grasped within the longevity research community. So I have been devoting a series of blog entries devoted to the science of this topic. This is the fourth such entry, as I continue to study the vast probably-relevant literature and learn new things.
For those of you who are completely new to this line of discussion I suggest you start by reading the less technical introductory blog entry YGA Introduction to the YOUNGING Series – Emerging Aging Reversal Strategies and Treatments. After that, the next thing I suggest you read is the substantive blog YGB YOUNGING1.0 – THE EMERGING AGING REVERSAL STRATEGY. Then you can read the blog entry just prior to this one YGC MORE ON YOUNGING1.0 – THE EMERGING AGING REVERSAL STRATEGY.
In these blog entries, I proposed at least three key hypotheses. The second of these is unique in the published literature related to longevity.
First, that humans and other advanced biological organisms enjoy in their adult years a number of mechanisms of systemic age reversal, these going on simultaneously with aging processes. I call these YOUNGING processes. While the existence of such mechanisms is acknowledged in the longevity research community, I think it is fair to observe that they are not being treated there now as very important. Evidence suggests that these processes range from ones imperceptible in their operation to long-term processes which can reverse many of the landmarks of aging to much younger states. These may require a decade or more for their operation.
Second, I have asserted that H3K27me2-3 histone demethylation via expression of JMJD3 is the fundamental basis for one or more key YOUNGING processes in advance organisms including humans.
Third, controlling and limiting chronic systematic inflammation is also necessary for essential YOUINGING processes to happen.
The purpose of this present blog entry is to further expand on these hypotheses, providing further science background and expressing them in deeper and more precise ways. In particular, my intent is to identify the biological mechanisms underlying the second assertion above. And, to provide published research evidence for each key step of my argument. In this discussion I will cover background related to Polychrome Repressive Complexes and highlight the importance of stem cells for rejuvenation. I will discuss some of the critical demethylase-dependent and demethylase-independent rules of JMJD3 for chromatin reorganization required to allow cell lineage differentiation. I dig further into on the the critical roles of histone methylation and JDJM3 in phases of aging, and discuss the companion roles of UTX with JMJD3. I will also discuss other age-related histone modifications including methylation of other histone sites besides H3K27me2-3 And I touch on other important epigenetic histone post-translational modifications including acetylation, phosphorylation, ubiquitylation, and sumoylation.
II. RECAP OF PREVIOUS BLOG ENTRIES IN THIS SERIES
Re. expression of JMJD3 and consequent repression of H3K27me2-3 is the basis for one or more key YOUNGING processes in humans.
Previous blog entries in this series support the key proposition that expression of JMJD3 and consequent repression of H3K27me2-3 play in the development in a wide variety of biological organisms, including us Homo Erectus. In short:
-
- Early development of organisms absolutely requires the turning on of a number of key growth and development genes, ones whose expression is otherwise inhibited by. H3K27me2-3 methylation which is inherited from parents, must be turned off early in development to enable expression of a wide variety of genes required for growth and development. The developing organism therefore inhibits H3K27me2-3 methylation via expression of JMJD3, allowing expression of those genes. Later, after reproductive maturity is achieved there is a rather abrupt period where the expression of JDJM3 is turned significantly down initiating adult aging. This two-step process appears to be very fundamental for most if not all species of living organisms. It is observed in insects and plants as well as vertebrates. A profound YOUNGING process is required for the development of advance organisms including humans. That process is normally dialed way down as childbearing years come to an end. This happens in part because of H3K27me2-3 histone methylation which dials down the activity the activities of numerous developmental genes.
- If we, much later in life, want to foster longevity associated with cell rejuvenation, we must discover means for turning complexes of the same genes back on in limited and controlled ways. I am suggesting that a best way to turn those genes back on as part of initiating a basic YOUNGING process is the same way they were originally turning on, by inhibiting H3K27me2-3 methylation via expression of JMJD3.
- Recall, that YOUNGING01 as we have characterizred it is a reverse epigenetic aging process in mature adults back towards an early adult stage such as may be experienced typically in someone’s mid 20s. It is definitely is not intended or purport to revert a human back to childhood or infancy as in The Curious Case of Benjamin Button A very important implication is that epigenetic age regression at a cellular level is a vital part of what is wanted and needed, but epigenetic regress all the way back to pluripotency (ie all the way back to stem cell level). is neither needed or wanted. Specifically, cellular regession must not go so far back that cell lineage markers are lost. Specifically, epigenetic age regression of adult somatic cells is needed to regress them back into precursor cells capable in turn of turning into young and vital cells in the same lineage.
III. AN UNDERLYING MECHANISM BEHIND THE YOUNGING 1.0 HYPOTHESIS
In summary, polychrome repressive complex (PRC2) hypermethylation of histones (H3K27me) increases with advanced age in stem cells, and is responsible for decreasing efficiency of stem cells to achieve their routine tasks of differentiating and replacing other senescent or dead somatic body cells. The result is that lifelong processes of cell renewal via stem cells slows down and is impaired. JDJM3 reverses the aging cell epigenetic signature due to PRC2 histone hypermethylation, particularly in HSC stem cells responsible for most body cell renewal. Couple this reversal of hyper methylation with improving NF-KB and NRF2 signaling and reducing ROS levels by controlling chronic inflammation, the result is that old stem cells behave a lot more lot younger ones – the YOUNGING 1.0 age reversal effect. I herein lay out the logical steps that lead to these conclusions. And I cite a variety of recent research results related to these steps.
- Body cells mature, die and are replaced by newly-differentiated stem cells on an ongoing process. Without this happening advanced organisms could not survive; life for us would be impossible. Red blood cells in humans, for example, have a lifespan of only about 4 months. Unlike other cell types, they cannot reproduce by cell division and must be replaced by differentiated stem cells. Essentially all cell types are ultimately subject to such replacement, although expected cell lifespans may vary radically by type.
- Of particular interest are hemopoietic stem cells (HSCs) which live in bone marrow niches and are responsible for replacement for all blood cell types, a process known as hematopoiesis. HSCs may be responsible for replacement of many other body cell types as well as blood-line ones.
- All stem cells themselves reproduce via normal cell division and have relatively long lifespans. However with advanced aging the structure and composition of stem cells change significantly and their ability to differentiate into their destination somatic cell types becomes compromised.
- This effect may just by itself explain many of the manifestations of advanced aging as organs become starved of healthy replacements for senescent or morbid cells.
- Among the known age-related impacts of HSCs are epigenetic shifts that significantly down regulate their accurate reproduction and differentiation into their designated body cell types. Included among these shift are A. Hyper histone methylation at the sites resulting from rising levels of PRC2, B Hypo histone acetylation at the H4K16 site. And C. Unrepaired or under-repaired DNA damage associated with cell reproduction.
- Some of the most significant aging related shifts in stem cells can be reversed by known interventions that affect the epigenome. One of these, highlighted in the YOUNGING 1.0 approach is global histone demethylation at the H3K27me2-3 sites via induction of the demethylase JMJD3. Among its effects, JMJD3 accelerates the differentiation of stem cells.
IV. MORE DETAILED DISCUSSION AND RESEARCH CITATIONS
- Body cells, whether of dividing types or not, mature, die and are replaced by newly-differentiated stem cells as an ongoing life-long process. About hemopoietic stem cells (HSCs)
The necessary replacement of normal body (somatic) cells by newly differentiated stem cells has been studied for some time. The natural renewal of blood cells is a case in example. From the 2017 publication On the Mechanism of Human Red Blood Cell Longevity: Roles of Calcium, the Sodium Pump, PIEZO1, and Gardos Channels, “In a healthy adult, the transport of O2 and CO2 between lungs and tissues is performed by about 2 · 1013 red blood cells, of which around 1.7 · 1011 are renewed every day, a turnover resulting from an average circulatory lifespan of about 120 days — Human red blood cells (RBCs) have a prescribed lifespan of about 4 months before being cleared from the circulation. During this period, RBCs devoid of organelles and biosynthetic capacity experience irreversible age-related changes in metabolism, membrane transport, ionic composition, cortical cytoskeleton, and immune-reactivity, among others (Beutler, 1985b; Clark, 1988; Romero and Romero, 1999; Lew et al., 2007; Tiffert et al., 2007; Lutz, 2012; Franco et al., 2013; Lutz and Bogdanova, 2013). Nevertheless, under the microscope, the appearance of RBCs from healthy adults remains remarkably uniform, regardless of cell age. — . That such manufacturing precision can be maintained by a bone marrow producing about 7 · 109 cells per hour in a normal adult is a truly remarkable feat of evolutionary bioautomation. And no less remarkable is the mechanism that enables the homeostatic stability of the RBCs throughout senescence. — We suggest that the need to maintain optimal circulatory performance has driven the evolution of a remarkably costefficient compensation strategy to preserve RBC volume and to extend RBC lifespan.”
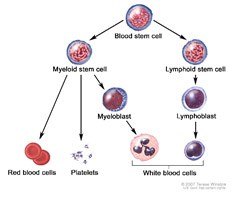
Constant renewal of all blood cell types depend on constant differation and regeneration of blood stem cells, long known as hematopoietic stem cells (HSCs). In fact research suggests that HSCs may in fact be the stem cell source of all the body’s cell types. The 2013 publication Hematopoietic Stem Cells Are Pluripotent and Not Just “Hematopoietic” reports “Over a decade ago, several preclinical transplantation studies suggested the striking concept of the tissue-reconstituting ability (often referred to as HSC plasticity) of hematopoietic stem cells (HSCs). While this heralded an exciting time of radically new therapies for disorders of many organs and tissues, the concept was soon mired in controversy and remained dormant for almost a decade. This commentary provides a concise review of evidence for HSC plasticity, including more recent findings based on single HSC transplantation in mouse and clinical transplantation studies. There is strong evidence for the concept that HSCs are pluripotent and are the source for the majority, if not all, of the cell types in our body. — Many tissues and organs in our body possess variable but significant regenerative capacity. Some organs such as bone marrow (BM), skin, and the mucosa of the gastrointestinal tract are characterized by life-long cell turnover. In these organs the mature cells have finite life spans and are continually replaced by more primitive progenitor cells. Others such as skeletal muscle and liver are characterized by limited cell turnover in the steady-state, but are capable of significant regeneration following tissue injury and loss of constituent cells. To account for the cell turnover in these organs, the concept of stem cells, cells that can self-renew and generate progeny committed to differentiation in specific pathways, emerged decades ago. Further, it was generally held that stem cells possess organ/tissue specificity; for example, hematopoietic stem cells (HSCs) generate only blood cells. — Against this long-held belief, striking tissue-reconstituting capability of HSCs (often referred to as HSC plasticity) was reported about a decade ago and suggested exciting new avenues of therapy for disorders of many organs and tissues. However, these reports were soon followed by others with negative results and papers offering different interpretations, as exemplified in the disputes among a number of major laboratories [1–3]. Our laboratory has also been engaged in the studies of HSC plasticity using single-cell HSC transplantation and has obtained unequivocal evidence for the HSC-origin of fibroblasts/myofibroblasts, adipocytes, and osteo-chodrocytes, major cell types comprising connective tissue. This commentary consists of a brief summary of our studies of connective tissue and of studies reported from other laboratories indicating an HSC-origin of other major cell types. We believe these findings strongly support the concept that HSCs are the source for the majority, if not all, of the cell types in our body.” This concept, of the capability of HSCs to differentiate into many if not all cell types has been widely picked up In the literature, as exemplified by these documents.
2. Many of the manifestations of advanced aging could be due simply to decline in differentiation and functionality of stem cells, HSCs in particular.
If stem cells can no longer function so as to satisfy ongoing needs for replacement of normal body cell types (somatic cells), many if not most of the normal symptoms of advanced aging might be explained by that fact alone. The 2007 publication Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation reports: “Age-related defects in stem cells can limit proper tissue maintenance and hence contribute to a shortened lifespan. Using highly purified hematopoietic stem cells from mice aged 2 to 21 mo, we demonstrate a deficit in function yet an increase in stem cell number with advancing age. Expression analysis of more than 14,000 genes identified 1,500 that were age-induced and 1,600 that were age-repressed. Genes associated with the stress response, inflammation, and protein aggregation dominated the up-regulated expression profile, while the down-regulated profile was marked by genes involved in the preservation of genomic integrity and chromatin remodeling. Many chromosomal regions showed coordinate loss of transcriptional regulation; an overall increase in transcriptional activity with age and inappropriate expression of genes normally regulated by epigenetic mechanisms was also observed. Hematopoietic stem cells from early-aging mice expressing a mutant p53 allele reveal that aging of stem cells can be uncoupled from aging at an organismal level. These studies show that hematopoietic stem cells are not protected from aging. Instead, loss of epigenetic regulation at the chromatin level may drive both functional attenuation of cells, as well as other manifestations of aging, including the increased propensity for neoplastic transformation.”
Also highly relevant in this regard is the 2015 publication Epigenetic Control of Stem Cell Potential during Homeostasis, Aging, and Disease “Stem cell decline is an important cellular driver of aging-associated pathophysiology in multiple tissues. Epigenetic regulation is central to establishing and maintaining stem cell function, and emerging evidence indicates that epigenetic dysregulation contributes to the altered potential of stem cells during aging. Unlike terminally differentiated cells, the impact of epigenetic dysregulation in stem cells is propagated beyond self; alterations can be heritably transmitted to differentiated progeny, in addition to being perpetuated and amplified within the stem cell pool through self-renewal divisions. — Tissue-specific stem cells are imbued with self-renewal potential and the capacity for differentiation to generate mature effector cells, and thus they are responsible for sustained function of tissues throughout life. Aging is associated with the progressive inability to maintain tissue homeostasis or robustly regenerate tissue after injury or stress. These processes are mediated by tissue-specific stem cells, suggesting that impaired stem cell function may underlie central cellular pathophysiologies associated with aging. Indeed, mounting evidence indicates that degenerative aging-associated changes in adult stem cells are a central driver of many age-related phenotypes (reviewed in Oh et al., 2014, Liu and Rando, 2011, Behrens et al., 2014, Rossi et al., 2008). The mechanistic basis for aging-associated stem cell decline is not completely understood, but numerous studies have shown that loss of polarity (Florian et al., 2012), mitochondrial dysfunction (Bratic and Larsson, 2013), altered autophagy (Warr et al., 2013), replicative stress (Flach et al., 2014), and accrual of DNA damage (Rossi et al., 2007, Rübe et al., 2011, Yahata et al., 2011, Wang et al., 2012, Beerman et al., 2014) all contribute to stem cell aging. In addition, increasing evidence suggests that epigenetic dysregulation is also an important mechanistic driver of stem cell aging.”
A central hypothesis being proposed in this blog entry is that restoring a more youthful profile of epigenetic regulation in HSC stem cells via global histone deacetylation of H3K27me2-3 via JDJM3 is a central mechanism of the age reversal processes I have called YOUNGING 1.0
Among its effects, JMJD3 accelerates the differentiation of stem cells. The 2016 publication Transient ectopic expression of the histone demethylase JMJD3 accelerates the differentiation of human pluripotent stem cells reports: “Harnessing epigenetic regulation is crucial for the efficient and proper differentiation of pluripotent stem cells (PSCs) into desired cell types. Histone H3 lysine 27 trimethylation (H3K27me3) functions as a barrier against cell differentiation through the suppression of developmental gene expression in PSCs. Here, we have generated human PSC (hPSC) lines in which genome-wide reduction of H3K27me3 can be induced by ectopic expression of the catalytic domain of the histone demethylase JMJD3 (called JMJD3c). We found that transient, forced demethylation of H3K27me3 alone triggers the upregulation of mesoendodermal genes, even when the culture conditions for the hPSCs are not changed. Furthermore, transient and forced expression of JMJD3c followed by the forced expression of lineage-defining transcription factors enabled the hPSCs to activate tissue-specific genes directly. We have also shown that the introduction of JMJD3c facilitates the differentiation of hPSCs into functional hepatic cells and skeletal muscle cells. These results suggest the utility of the direct manipulation of epigenomes for generating desired cell types from hPSCs for cell transplantation therapy and platforms for drug screenings.
- The histone code, the epigenetic regulation of stem cells, and the interplay between epigenetic modifications
There is increasing recognition in the research community of the importance of epigenetic histone modifications for the global activation and in the activation of families of genes. And, of the profound importance of the gene-regulating impact of such modifications in both early developments and advanced aging. “Such modifications include acetylation, methylation, phosphorylation and ubiquitination The functional consequences of histone modifications can be direct, causing structural changes to chromatin, or indirect, acting through the recruitment of effector proteins. All histone modifications are removable, which may therefore provide a flexible way for regulation of gene expression(ref).”
Further, there can be significant interaction between methylation, acetylation, and ubiquititination processes, as pointed out in the document Epigenetic interplay of histone modifications and DNA methylation mediated by HDA6. “Our studies demonstrate that HDA6 integrates DNA methylation and histone modifications in gene silencing by interacting with MET1 and FLD (Fig. 1). More recently, OTU6 was found to directly interact with the histone lysine demethylase LDL1/KDM1C in plants,25 suggesting that regulatory crosstalk between histone demethylation and deubiquitination through the direct interaction between LDL1/KDM1C and OTU6. Taken together, data described above indicate that the crosstalk among different epigenetic modifications could be mediated by the interaction of various histone modification proteins and complexes.” This diagram from that publication shows how a histone deacetylase can lead to an altered methylation profile.
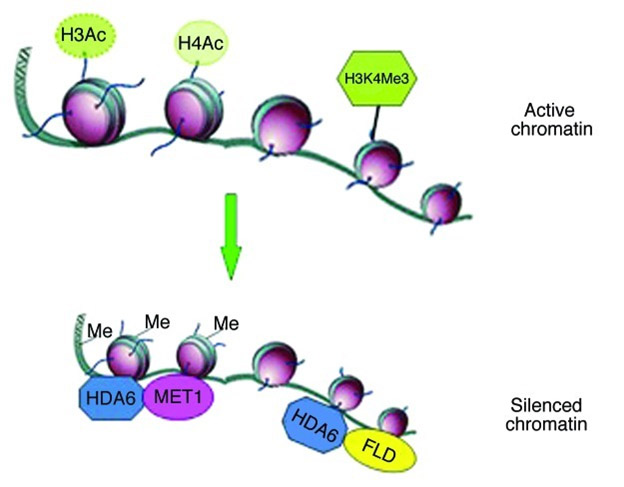
”Gene silencing mediated by HDA6. The gene activation markers, H3 acetylation, H4 acetylation and H3K4 tri-methylation are associated with active chromatin. HDA6 can mediate gene silencing through interacting with MET1 and FLD, resulting in DNA methylation, histone deacetylation and histone demethylation.”
4. With advanced aging the structure and composition of stem cells change significantly and their ability to differentiate into their destination somatic cell types becomes compromised
This theme, for example is elaborated within the 2020 research publication Understanding intrinsic hematopoietic stem cell aging. “Aging is the largest risk factor for many chronic diseases and disabilities. Not surprisingly, aging is also the major risk factor for several hematologic syndromes and malignancies, such as myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML).1 Moreover, aging has a negative impact on HSC regenerative capacity, and for this reason, cell-intrinsic mechanisms of aging are important putative targets for therapeutic interventions in order to ameliorate the consequences of aging on HSC and on the hematopoietic system.2 Understanding the mechanisms of HSC aging will provide the scientific community with new tools to improve the regenerative capacity of healthy HSC and thus the function of the hematopoietic system in the elderly.” I have found the following diagram from that publication to be particularly helpful in illustrating the multiple cell-specific aspects of aging stem cells.
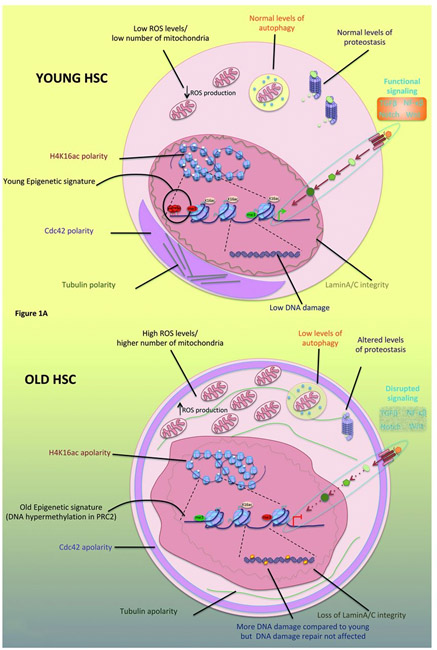
Off particular interest in the present discussion is histone hypermethylation due to increasing PRC2 expression, which can be reversed using the JDJM3 histone 27 decatylase. Other impacts of aging shown in the diagram include “Increase in phenotypic HSC number and decrease regenerative capacity, Myeloid skewing, DNA damage, Clonality, Epigenetic drift, Cell polarity, Metabolic alterations and impaired autophagy, Altered proteostasis, Alterations in intrinsic signaling pathways”
This following diagram compares histone acetylation/deacetylation impacts at the H4K16 position in plants as well as in mammals, illustrating how the impacts if ancient histone modifications have been evolutionarily conserved across plant and animal species.
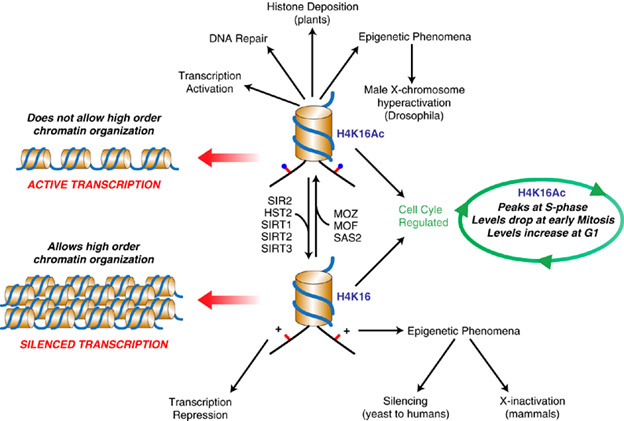
Image source “H4K16 versus H4K16Ac. The acetylation status of lysine 16 in histone H4 (represented as a blue circle) has a critical role in multiple functions in chromatin regulation throughout evolution as shown. Whereas acetylation of K16 is involved in active transcription and open chromatin, non-acetylated K16 is associated with gene silencing and heterochromatin compacted structure.”
5. Underlying HSC stem cell aging and its negative effects are alterations in the DNA methylation landscape resultant from the proliferation history of the HSCs
The 2014 publication Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging reports “The functional potential of hematopoietic stem cells (HSCs) declines during aging, and in doing so, significantly contributes to hematopoietic pathophysiology in the elderly. To explore the relationship between age-associated HSC decline and the epigenome, we examined global DNA methylation of HSCs during ontogeny in combination with functional analysis. Although the DNA methylome is generally stable during aging, site-specific alterations of DNA methylation occur at genomic regions associated with hematopoietic lineage potential and selectively target genes expressed in downstream progenitor and effector cells. We found that age-associated HSC decline, replicative limits, and DNA methylation are largely dependent on the proliferative history of HSCs, yet appear to be telomere-length independent. Physiological aging and experimentally enforced proliferation of HSCs both led to DNA hypermethylation of genes regulated by Polycomb Repressive Complex 2. Our results provide evidence that epigenomic alterations of the DNA methylation landscape contribute to the functional decline of HSCs during aging.”
V. ADDITIONAL CHARACTERISTICS OF H3K27me2-3 HISTONE METHYLATION, POLYCHROME REPRESSIVE COMPLEXES, AND JDJM3
Basic to our YOUNGING1 hypothysis is the proposition that the age reversal process involves the global demythelation of doubly or triply methylated lysine 27 on histone 3, that is of H3K27me2-3. We have suggested that YOUNGING in mature adults can be triggered by upgrading expression of a significant demethylase for H3K27me2-3, of which there are two, known as JDJM3 and UTX, What typically happens is that there is a lifelong dance of double and triple methylation at this histone site and demethylation. To start, what initiates such methylation?
- Introducing the Polycomb Repressive Complex2 (PRC2). The 2020 publication Methylation of H3K27me2-3 is induced by the specific methylase PRC2: Post-translational modifications of PRC2: signals directing its activity. “Polycomb repressive complex 2 (PRC2) is a chromatin-modifying enzyme that catalyses the methylation of histone H3 at lysine 27 (H3K27me1/2/3). This complex maintains gene transcriptional repression and plays an essential role in the maintenance of cellular identity as well as normal organismal development. The activity of PRC2, including its genomic targeting and catalytic activity, is controlled by various signals. Recent studies have revealed that these signals involve cis chromatin features, PRC2 facultative subunits and post-translational modifications (PTMs) of PRC2 subunits. Overall, these findings have provided insight into the biochemical signals directing PRC2 function, although many mysteries remain.”
- JMJD3 plays a critical role in this lifelong dance of aging methylation and demethylation. This is reflected in the title of the 2014 publication Histone demethylase Jumonji D3 (JMJD3/KDM6B) at the nexus of epigenetic regulation of inflammation and the aging process. “Histone methylation is involved in the epigenetic control of immune responses and cellular senescence. Jumonji domain-containing protein 3 (JMJD3), also called lysine-specific demethylase 6B (KDM6b), is an inducible histone demethylase which enhances immune responses and can trigger cellular senescence. JMJD3 potentiates gene expression by demethylating repressive H3K27me3 epigenetic marks in promoters and gene bodies. Moreover, JMJD3 also stimulates transcription in a demethylase-independent manner by mediating interactions between chromatin modifiers. JMJD3 can enhance both pro-inflammatory and anti-inflammatory responses by targeting distinct transcription factors in a context-dependent manner in gene promoters. For instance, JMJD3 can induce macrophage M2 polarization via STAT6 signaling. JMJD3 also interacts with T-bet factor and induces Th1 differentiation of CD4(+) T cells. Moreover, JMJD3 can activate TGF-β signaling through the SMAD3 pathway. Conversely, JMJD3 displaces polycomb complexes from the INK4 box, which induces the expression of INK4a and triggers cellular senescence. JMJD3 can also enhance the nuclear localization of p53 and thus regulate its function. The control of INK4 box and p53 is closely related to the regulation of the aging process. We will briefly review the inducible properties of JMJD3 expression and then focus on the role of JMJD3 in the regulation of inflammation and senescence through different signaling pathways. We emphasize that an inflammatory milieu and cellular stress can enhance immune responses and provoke cellular senescence via epigenetic regulation through JMJD3 activation.”
- JMJD3 and early development. From the 2015 publication JMJD3 as an epigenetic regulator in development and disease “— 1.1. JMJD3 and development Embryonic stem cells (ESCs) repress developmental genes by utilizing H3K27 trimethylation, but ESC deficiency in JMJD3 does not seem to affect stem cell maintenance and self-renewal capacity (Mansour et al., 2012; Ohtani et al., 2013). During differentiation, H3K27 methylation is removed in a tissue- and cell-specific manner, and the demethylases, JMJD3 and UTX, are directly involved in embryogenesis into the three germ layers, endoderm, mesoderm, and ectoderm, of a developing vertebrate. — 1.1.1. Endoderm JMJD3 and UTX drive the formation of the germ layer, endoderm, which gives rise to the gastrointestinal tract, respiratory tract, endocrine glands, and the auditory and urinary systems. Endoderm commitment is controlled by the WNT signaling pathway. — 1.1.2. Mesoderm JMJD3 and UTX are also involved in the formation of the germ layer, mesoderm, but JMJD3 can partially compensate for the loss of UTX during ESC differentiation into mesoderm (Morales Torres et al., 2013). Mesodermal development leads to the formation of muscle tissue, spleen, cartilage, bone, skin, kidneys, gonads, heart, blood vessels, and blood cells. Jmjd3 deficiency in ESCs significantly increases repressive H3K27me3 marks on the promoter of the mesodermal regulator, Brachyury and decreases its expression, leading to impaired recruitment of β-catenin (Ohtani et al., 2013), which is a prerequisite for WNT-induced mesoderm differentiation. During late mesoderm differentiation, JMJD3 is also essential for normal organ development. Jmjd3 knockout in ESCs reduces endothelial cell differentiation as well as cardiac progenitor cell differentiation (Ohtani et al., 2013). In addition, Jmjd3 knockout mice at E17.5 demonstrate impaired spleen development, with smaller size and hyperemic areas (Li, 2014a). Furthermore, JMJD3 affects bone formation. Endochondral bone formation and ossification begins with multipotent mesenchymal stem cell (MSC) differentiation into chondrocytes. This cartilage maturation during endochondral bone formation is regulated by JMJD3 and its association with the transcription factor, RUNX2, promoting proliferation and hypertrophy of chondrocytes (Zhang et al., 2015). JMJD3 can also direct MSCs to differentiate preferentially into one lineage over another lineage. — 1.1.3. Ectoderm Formation of the germ layer, ectoderm, is also regulated by JMJD3 and partially by UTX (Morales Torres et al., 2013). The ectoderm can differentiate into the nervous system, including the spine, peripheral nerves, and brain. Key regulators in neurogenesis, Pax6, Sox1, and Nestin, harbor bivalent marks, H3K4me3/H3K27me3, which are dynamically regulated during differentiation, and Jmjd3 can directly regulate expression of these key regulators in neurogenesis (Burgold et al., 2008). Jmjd3 is also a key regulator in Shh-dependent neural tube development (Shi et al., 2014). In fact, Jmjd3 is essential for ESC commitment into neural lineages. In the developing spinal cord, trimethylation of H3K27 also regulates BMP activity, which, in turn, leads to JMJD3 interaction with the transcription factors, SMAD1/4, to activate the notochord-derived BMP antagonist, Noggin (Akizu et al., 2010) (Fig. 1C). This negative feedback loop ensures rigorous and anatomically defined spinal cord formation. —. Overall, the important role of JMJD3 in the formation of all three germ layers suggests that JMJD3 is fundamental in cell fate and plasticity.”
- JDMJ3 is modulated by the polychrome repressive complex so as to control a network of genes related to puberty. A part of the dance. Recalling that KDM6b is the gene expressing JMJD3, the 2021 publication Polycomb represses a gene network controlling puberty via modulation of histone demethylase Kdm6b expression tells this story: “Female puberty is subject to Polycomb Group (PcG)-dependent transcriptional repression. Kiss1, a puberty-activating gene, is a key target of this silencing mechanism. Using a gain-of-function approach and a systems biology strategy we now show that EED, an essential PcG component, acts in the arcuate nucleus of the hypothalamus to alter the functional organization of a gene network involved in the stimulatory control of puberty. A central node of this network is Kdm6b, which encodes an enzyme that erases the PcG-dependent histone modification H3K27me3. Kiss1 is a first neighbor in the network; genes encoding glutamatergic receptors and potassium channels are second neighbors. By repressing Kdm6b expression, EED increases H3K27me3 abundance at these gene promoters, reducing gene expression throughout a gene network controlling puberty activation. These results indicate that Kdm6b repression is a basic mechanism used by PcG to modulate the biological output of puberty-activating gene networks.”
JDJM3 and H3K27me3 histone methylation play mutiple roles in lifelong normal and advanced aging as exemplified by the following discussions of tissue repair, the longevity factors KLOTHO and foxo3a.
- JMJD3 and lifelong tissue repair From the 2016 publication JMJD3 as an epigenetic regulator in development and disease “1.2. JMJD3 and cell plasticity Not only does JMJD3 play a role in cellular differentiation in the developing embryo, it regulates cellular processes in differentiated tissues. Specifically, Jmjd3 is intimately involved in tissue repair. Following bone injury, osteoclasts are responsible for bone resorption, and abnormal osteoclast differentiation can lead to osteoporosis. RANKL stimulation of the osteoclast cell surface receptor, RANK, leads to osteoclast differentiation. This is accompanied by JMJD3-mediated demethylation of H3K27me3 at the Nfatc1 gene, a gene responsible for bone mass (an). These studies suggest that JMJD3 mediates osteoclast differentiation after bone injury and may limit the onset of osteoporosis. JMJD3 has also been implicated in skin repair (Shaw and Martin, 2009). Whereas Polycomb genes, which are involved in histone methylation, are significantly downregulated during murine skin repair, the demethylases, Jmjd3 and Utx, are markedly upregulated, leading to less Polycomb-mediated silencing of the wound repair genes, Myc and Egfr (Shaw and Martin, 2009). Whether JMJD3 is directly involved in other types of tissue repair is not yet known, but JMJD3 is likely involved in the cellular plasticity involved in tissue repair (ref).”
- “In fact, JMJD3 may play a direct role in the reprogramming of adult cells into a pluripotent state. The ability to revert differentiated cells back into a pluripotent, embryonic stem cell-like state has been a breakthrough for patient-specific disease modeling and drug testing. This cellular reprogramming is induced by the exogenous addition of the transcription factors, Sox2, Oct4, c-Myc, and Klf4. OCT4 is critical for generating induced pluripotent stem cells (iPSCs) and maintaining pluripotency (Lowry et al., 2008; Pesce and Scholer, 2001). Activation of OCT4 occurs in parallel with the recruitment of JMJD3 to chromatin, suggesting that JMJD3 is involved in cellular reprogramming (Apostolou and Hochedlinger, 2013). Our own research has shown that JMJD3 is a potent negative regulator of cellular reprogramming (Zhao et al., 2013) (Fig. 4). Ablation of Jmjd3 in mouse embryonic fibroblasts increases iPSC formation, whereas, ectopic Jmjd3 expression inhibits reprogramming by both histone demethylase-dependent and -independent mechanisms (Zhao et al., 2013). Further understanding of the role of JMJD3 in cellular reprogramming and tissue repair may lead to therapeutic test-beds of iPSCs and enhance wound repair(ref).”
- One mechanism of aging is down-regulation of the longevity proteins klotho and foxo3a consequent to increasing h3k27me3 with aging. At least this is observed in mouse kidney cells. The 2020 publication Epigenetic Regulation of KL (Klotho) via H3K27me3 (Histone 3 Lysine [K] 27 Trimethylation) in Renal Tubule Cells reports: “KL (klotho) levels decline with age, which is an important mechanistic driver of aging. KL gene deficiency is associated with hypertension. The purpose of this study is to investigate the potential role of H3K27me3 (histone 3 lysine [K] 27 trimethylation) in the regulation of KL gene expression and examine the related molecular pathways that may drive kidney cell aging. Kidneys were collected from 6-month-old WT (wild type; young WT), 30-month-old WT (aged WT), and 6- (young) and 20-month-old (aged) KL mutant mice, respectively. We demonstrated that the H3K27me3 level was increased in kidneys of aged WT and KL mutant mice versus young WT mice. Elevation of H3K27me3 levels was likely due to downregulation of the H3K27 (histone H3 Lys 27)-specific demethylase JMJD3 (the Jumonji domain containing-3) in the aged kidneys. Inhibition of PRC2 (polycomb repressive complex C2; histone trimethyltransferase) decreased the H3K27me3 levels leading to an increase in the expression of KL in cultured primary renal tubule cells assessed by Western blot and KL promoter activity assays. The chromatin immunoprecipitation qPCR assay revealed that H3K27me3 was physically associated with the KL promoter region. Furthermore, aging impaired the SGK1 (serum- and glucocorticoid-induced protein kinase 1)/FOXO3a (the forkhead box class O 3a) signaling leading to upregulation of p53 and p16 (aging markers) in the kidney of aged WT mice. KL may regulate the SGK1/FOXO3 signaling, which was decreased due to KL deficiency. Thus, aging-associated downregulation of KL gene expression may be partly attributed to upregulation of H3K27me3 levels. Downregulation of KL may impair the SGK1/FOXO3 signaling contributing to kidney cell aging.” Of course. The YOUNGING01 response to this situation is downregulation of H3K27me3 via JDJM3.
- JMJD3 is recruited by the Yamanaka pluripotency factor KLF4. As reported in the 2020 publiction JMJD3 acts in tandem with KLF4 to facilitate reprogramming to pluripotency “The interplay between the Yamanaka factors (OCT4, SOX2, KLF4 and c-MYC) and transcriptional/epigenetic co-regulators in somatic cell reprogramming is incompletely understood. Here, we demonstrate that the histone H3 lysine 27 trimethylation (H3K27me3) demethylase JMJD3 plays conflicting roles in mouse reprogramming. y JMJD3 also promotes enhancer-promoter looping through the cohesin loading factor NIPBL and ultimately transcriptional elongation. This competition of forces can be shifted towards improved reprogramming by using early passage fibroblasts or boosting JMJD3’s catalytic activity with vitamin C. Our work, thus, establishes a multifaceted role for JMJD3, placing it as a key partner of KLF4 and a scaffold that assists chromatin interactions and activates gene transcription.” Note here the interesting observation that JMJD3 “induces the pro-senescence factor Ink4a” which would seem to lead cells down the path to senescence, the opposite direction from regression to “stemness” and pleuripotency. True. But only a problem if we fall into the trap of thinking of all similar cells in an organ as doing the same thing. As I have pointed out, different cells in an organ can healthily be in different epigenetic states, most in a mature but healthily functioning state, some in senescence, and sone in epigenetic regression, i.e. undergoing YOUNGING. Recall that the Yamanaka OSKM cell-level YOUNGING journey to pluripotency absolutely requires senescence signaling to trigger it. So the fact that a non-histone related impact of JMJD3 promotes cell senescence is part of its action which fosters cell-level reprogramming to pluripotency.
- JDJM3 expression can be dysregulated so as to induce inflammatory signaling in the case of alcohol dependence. The 2019 publication Dysregulation of the histone demethylase KDM6B in alcohol dependence is associated with epigenetic regulation of inflammatory signaling pathways reports “Through quantitative mRNA expression analysis of over 100 epigenetic enzymes, we identified 11 that are dysregulated in the brain of alcohol dependent rats. The histone 3, lysine 27 (H3K27me3) demethylase KDM6B, was found to have region‐specific dysregulation in the prefrontal cortex and nucleus accumbens and also in human alcoholic brain tissues. A ChIP‐sequencing analysis showed that alcohol‐induced changes in H3K27me3 were enriched at genes in the IL‐6 signaling pathway, thereby implicating a novel KDM6B‐mediated epigenetic signaling pathway integrated with inflammatory signaling pathways that underlie alcohol addiction.”
- JDMJ3 plays an important role in cancer processes. In this respect it is like many other substances involved in growth and development such as IGF1 and OCT4. The 2016 publication The roles of histone demethylase UTX and JMJD3 (KDM6B) in cancers: Current progress and future perspectives reports: “Aberrant epigenetic reprogramming occurs frequently in the development of tumors. Histone H3 lysine 27 trimethylation (H3K27me3) exerts a repressive epigenetic mark on a large number of genes. UTX and JMJD3 are the only two histone demethylases which activate gene expression via demethylating H3K27me3 to H3K27me2 or H3K27me1. Current studies show that dysregulation of these two proteins are heavily linked to oncogenesis in various tissue types. Accumulating evidence suggested that there is remarkable therapeutic potential of targeting JMJD3 or UTX in different types of cancer. Herein, we shall give a brief review on the functional roles of JMJD3 and UTX in cancers and evaluate the available compounds and agents targeting UTX and JMJD3. Finally, we also discuss the several modalities that target UTX and JMJD3 for cancer therapy. This review will help to develop novel strategies to abolish or restore effects of UTX and JMJD3 in the pathogenesis of cancer.”
This is the published list of Highlights for the 2016 publication Histone demethylase JMJD3 at the intersection of cellular senescence and cancer
-
-
- Different studies demonstrate JMJD3 as a tumor suppressor and oncogene.
- Tumor suppressor pathways controlled by JMJD3 help to coordinate growth arrest.
- JMJD3 activates the senescence-associated secretory phenotype (SASP) observed in aging.
- Senescent-like cancer cells undermine normal tissue
-
This dual role of JMJD3 suggests that it’s activation for longevity purposes be done in a carefully regulated way.
VI. AGING INFLAMMATION AND STEM CELL RENEWAL PROCESSES
Regular readers of this blog by now know the importance I assign to control of chronic inflammation for health in advanced aging, and for the possibility of living a very long life. You know I have written a number of blog entries related to this topic and in fact have created a dietary supplement for the control of chronic inflammation. Related to this current blog entry, there appears to be strong evidence that control of chronic inflammation is critical for maintenance of healthy stem cell reproduction and hematopoiesis. A number of recent research publications have focused on this. One of these is Inflamm-Aging of hematopoiesis, Hematopoietic stem Cells and the Bone Marrow Microenvironment. ”All hematopoietic and immune cells are continuously generated by hematopoietic stem cells (HSCs) and hematopoietic progenitor cells (HPCs) through highly organized process of stepwise lineage commitment. In the steady state, HSCs are mostly quiescent, while HPCs are actively proliferating and contributing to daily hematopoiesis. In response to hematopoietic challenges, e.g., life-threatening blood loss, infection, and inflammation, HSCs can be activated to proliferate and engage in blood formation. The HSC activation induced by hematopoietic demand is mediated by direct or indirect sensing mechanisms involving pattern recognition receptors or cytokine/chemokine receptors. In contrast to the hematopoietic challenges with obvious clinical symptoms, how the aging process, which involves low-grade chronic inflammation, impacts hematopoiesis remains undefined. Herein, we summarize recent findings pertaining to functional alternations of hematopoiesis, HSCs, and the bone marrow (BM) microenvironment during the processes of aging and inflammation and highlight some common cellular and molecular changes during the processes that influence hematopoiesis and its cells of origin, HSCs and HPCs, as well as the BM microenvironment. We also discuss how age-dependent alterations of the immune system lead to subclinical inflammatory states and how inflammatory signaling might be involved in hematopoietic aging. Our aim is to present evidence supporting the concept of “Inflamm-Aging,” or inflammation-associated aging of hematopoiesis.”
Also relevant is the January 2021 review publication Inflammation and hematopoietic stem cells aging. “Hematopoietic stem cells (HSCs) replenish all lineages of blood cells throughout the lifespan. During aging, the repopulation capacity of HSCs declined, and aged HSCs display a tendency for myeloid differentiation. Several intrinsic and extrinsic factors have been identified to promote HSCs aging. In this review, we focus on the contribution of aging-associated inflammation in provoking HSCs aging and discuss the future research direction of inflammation and HSC aging.”
VII. YOUNGING PROCESSES AT A CELLULAR LEVEL CAN INVOLVE DEMETHYLATION OF DOUBLE OR TRIPPLE METHYLATED HISTONE LYSINES IN ADDITION TO OR BESIDES LYSINE 27
While I have focused specifically on H3K27me2-3 methylation up to this point, YOUNGING processes may require demethylation of other histone lysines such as H3K9me3. This is discussed for example in the following publications:
- The 2015 publication Histone Demethylase Expression Enhances Human Somatic Cell Nuclear Transfer Efficiency and Promotes Derivation of Pluripotent Stem Cells is another supporting the role of demethylation of trimethylated histones for cell reprogramming to younger more pluripotent states, this time related to lysine 9 instead of 27. “The extremely low efficiency of human embryonic stem cell (hESC) derivation using somatic cell nuclear transfer (SCNT) limits its potential application. Blastocyst formation from human SCNT embryos occurs at a low rate and with only some oocyte donors. We previously showed in mice that reduction of histone H3 lysine 9 trimethylation (H3K9me3) through ectopic expression of the H3K9me3 demethylase Kdm4d greatly improves SCNT embryo development. Here we show that overexpression of a related H3K9me3 demethylase KDM4A improves human SCNT, and that, as in mice, H3K9me3 in the human somatic cell genome is an SCNT reprogramming barrier. Overexpression of KDM4A significantly improves the blastocyst formation rate in human SCNT embryos by facilitating transcriptional reprogramming, allowing efficient derivation of SCNT-derived ESCs using adult Age-related Macular Degeneration (AMD) patient somatic nuclei donors. This conserved mechanistic insight has potential applications for improving SCNT in a variety of contexts, including regenerative medicine.”
- This closely related publication The Histone Demethylases Jhdm1a/1b Enhance Somatic Cell Reprogramming in a Vitamin-C-Dependent Manner relates a similar theme, this time involving trimethylation of histone lysine36. “Reprogramming of somatic cells into induced pluripotent stem cells (iPSCs) resets the epigenome to an embryonic-like state. Vitamin C enhances the reprogramming process, but the underlying mechanisms are unclear. Here we show that the histone demethylases Jhdm1a/1b are key effectors of somatic cell reprogramming downstream of vitamin C. We first observed that vitamin C induces H3K36me2/3 demethylation in mouse embryonic fibroblasts in culture and during reprogramming. We then identified Jhdm1a/1b, two known vitaminC-dependent H3K36 demethylases, as potent regulators of reprogramming through gain- and loss-offunction approaches. Furthermore, we found that Jhdm1b accelerates cell cycle progression and suppresses cell senescence during reprogramming by repressing the Ink4/Arf locus. Jhdm1b also cooperates with Oct4 to activate the microRNA cluster 302/367, an integral component of the pluripotency machinery. Our results therefore reveal a role for H3K36me2/3 in cell fate determination and establish a link between histone demethylases and vitamin C induced reprogramming.”
This publication also highlights a couple of other important factors: first that different histone lysine positions require different but closely related demethylases. E.g. demethylating H3K36me2/3 takes place via JHDM1a/1b while, as we have previously discussed, how demethylating H3K27me2/3 takes place via JDJM3. So, positioning YOUNGING as involving only H3K27me2/3 and JDJM3 has been an over-simplification. Second, that vitamin C is an important factor as far as cell-level YOUNGING is concerned. I have pointed this out in earlier blog entries. For example I devoted a section to discussing this topic mentioning additional literature citations and a diagram in the blog entry YGC More on YOUNGING1.0 – the emerging aging reversal strategy. A practical consequential inference is that any practical approach to initiating YOUNGING might benefit from generous self-dosage with vitamin C or actually require vitamin C to be effective. Please note that I am not at this point suggesting that vitamin C by itself is sufficient to initiate YOUNGING as I may hae hoped decades ago.
VIII. IS THE WHOLE YOUNGING 01 HYPOTHESIS SO RADICAL AS TO BE OUT OF STEP WITH MAINLINE SCIENCE?
I have to finally comment on this because I have learned that intelligent educated people may think that I have gone a bit off my rocker when I tell them that significant age reversal is now possible, that I am starting to understand how it works, that in fact I am doing it, and that it is likely working for me. I have discovered the secret of the Fountain of Youth? Me? Right now after so many thousands of years of failure to do so in the course of human history? Give me a break.
Most people with this reaction don’t know enough about biology or longevity to discuss the topic intelligently. So they mainly seek to change the subject, politely of course. On the other hand there is a growing number of key longevity scientists who believe like I do that aging is optional, like David Sinclair who wrote the seminal book Lifespan: Why We Age―and Why We Don’t Have To.
I do not believe the YOUNGING concept is radical, although it would have been so only only 5 years ago. Researchers are increasingly believing in the possibility of age-reversal via epigenetic reprogramming. By 2017, publications where appearing with titles like The Emerging View of Aging as a Reversible Epigenetic Process. As time rolls by, there have been more and more of them. A latest is the April 2021 publication A ride through the epigenetic landscape: aging reversal by reprogramming.
Another quite relevant April 2021 publication is Potential reversal of epigenetic age using a diet and lifestyle intervention: a pilot randomized clinical trial. This paper describes a small clinical trial that demonstrates limited epigenetic epigenetic age reversal age reversal; The endpoint of the study is epigenetic age as measured by a Horvath DNA methylation clock. +“– Herein we report on a randomized controlled clinical trial conducted among 43 healthy adult males between the ages of 50-72. The 8-week treatment program included diet, sleep, exercise and relaxation guidance, and supplemental probiotics and phytonutrients. The control group received no intervention. Genome-wide DNA methylation analysis was conducted on saliva samples using the Illumina Methylation Epic Array and DNAmAge was calculated using the online Horvath DNAmAge clock (2013). The diet and lifestyle treatment was associated with a 3.23 years decrease in DNAmAge compared with controls (p=0.018). DNAmAge of those in the treatment group decreased by an average 1.96 years by the end of the program compared to the same individuals at the beginning with a strong trend towards significance (p=0.066). Changes in blood biomarkers were significant for mean serum 5-methyltetrahydrofolate (+15%, p=0.004) and mean triglycerides (-25%, p=0.009). To our knowledge, this is the first randomized controlled study to suggest that specific diet and lifestyle interventions may reverse Horvath DNAmAge (2013) epigenetic aging in healthy adult males. Larger-scale and longer duration clinical trials are needed to confirm these findings, as well as investigation in other human populations. — Manipulations to slow biological aging and extend healthspan are of interest given the societal and healthcare costs of our aging population. Herein we report on a randomized controlled clinical trial conducted among 43 healthy adult males between the ages of 50-72. The 8-week treatment program included diet, sleep, exercise and relaxation guidance, and supplemental probiotics and phytonutrients. The control group received no intervention. Genome-wide DNA methylation analysis was conducted on saliva samples using the Illumina Methylation Epic Array and DNAmAge was calculated using the online Horvath DNAmAge clock (2013). The diet and lifestyle treatment was associated with a 3.23 years decrease in DNAmAge compared with controls (p=0.018). DNAmAge of those in the treatment group decreased by an average 1.96 years by the end of the program compared to the same individuals at the beginning with a strong trend towards significance (p=0.066). Changes in blood biomarkers were significant for mean serum 5-methyltetrahydrofolate (+15%, p=0.004) and mean triglycerides (-25%, p=0.009). To our knowledge, this is the first randomized controlled study to suggest that specific diet and lifestyle interventions may reverse Horvath DNAmAge (2013) epigenetic aging in healthy adult males. Larger-scale and longer duration clinical trials are needed to confirm these findings, as well as investigation in other human populations.”
This current blog series about YOUNGING is intended to be a meaningful contribution to this stream.
My current intention is that Part 5 of this YOUNGING series will be about practical ways for inducing YOUNGING 01, including combinations of activating JDJM3 and limiting chronic inflammation.

Pingback: YGC MORE ON YOUNGING1.0 – THE EMERGING AGING REVERSAL STRATEGY - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™
Pingback: YGB YOUNGING1.0 – THE EMERGING AGING REVERSAL STRATEGY - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™