
By James P. Watson with contributions and editing by Vince Giuliano
Introduction
(By Vince Giuliano)
Jim Watson and I have held that aging and biology, like the universe itself, have no centers. Like the universe, they can be looked at from many different frames of reference (viewpoints). And like for the universe what you see depends on your frame of reference. Different frames of reference may reveal different insights. For example, we have published a series of blog entries from the viewpoint of the telomere end of the chromosome (ref)(ref)(ref).
Why is DNA methylation important?
This blog entry looks at at aging, health and disease from the viewpoint of DNA methylation, that is, lifelong changes in methylation status of selected genes. This is a highly useful viewpoint because it:
- provides a theory of aging and a fairly concise definition of aging,
- provides the most concise measures of human aging we have, and
- is a good predictor of all-cause mortality.
According to this viewpoint, genomic methylation:
- plays crucial lifelong roles in human development and maturation, from embryogenesis right up to death,
- is one of the three major epigenetic mechanisms for gene activation and silencing,
- is a major causal factor in the program of aging,
- provides an explanation of how chronic inflammation accelerates the aging program,
- explains several other basic known mechanisms and effects of aging; E.g. loss of border proteins that protect CpG islands from methylation, and the fact that men age faster than women, and why women’s breasts age faster than the rest of their bodies,
- provides insights into the programs of aging that suggest possible hacks on those programs,
- has molecular mechanisms which are fairly well understood and well documented,
- helps explain a number of disease processes and disease susceptibilities,e.g. how hypermethyltion of the BRCA1 gene confers susceptibility to breast cancer,
- helps to explain how chronic stress leads to accelerated aging,
- plays a direct role in the pathogenesis of diabetes, cancer, other diseases,
- with aging, increases expression of repetitive DNA sequences and human endogenous retroviruses so as to lead to chromosomal/genomic instability, telomere attrition, and aneuploidy,
- helps explain selective hereditability of traits, and
- reveals important links to a number of other important topics and viewpoints related to health and aging we have discussed over the years such as other aspects of epigenetics, events in the NAD world, stress and hormesis, oxidative stress, importance of circadian rhythms, transposable elements and alternative splicing.
This blog entry is basically concerned with what DNA methylation is, its main properties and why it is important. We start with SECTION A – SOME DNA BASICS. If you know about DNA and the rudiments of genetics, you can possibly skip over this. SECTION B – DNA METHYLATION IS THE BEST CLOCK FOR AGING telegraphs a main reason for importance of this topic. The remainder of this entry SECTION C, D and E consist of more detailed information on facets and implications of DNA methylation organized according to numbered points written by Jim Watson. The concluding SECTION F briefly suggests a few possible practical health and longevity interventions based on the science discussed in this blog entry.
SECTION A – SOME DNA BASICS
(This Section put together by Vince Giuliano)
- How is DNA organized?
As a reminder, DNA is organized in a double-helix sugar-phosphate backbone joined by pairs of four base substances as shown in this diagram: Adenine, Thymine, Guanine and Cytosine.

2. Methylation
Methylation occurs when a methyl chemical group is attached to a Cytosine element in a base pair. The methylation of cytosine residues found directly adjacent to guanine elements in DNA is a fundamental feature of heterochromatin (tightly packed DNA; plays a role in the expression of genes) and serves as a method of gene regulation in euchromatin (” a lightly packed form of chromatin (DNA, RNA and protein) that is enriched in genes, and is often (but not always) under active transcription) as well.”
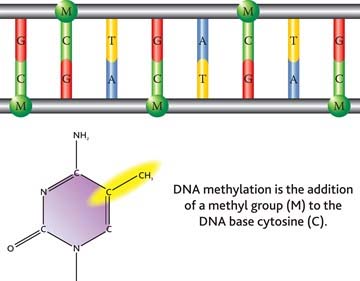
In the human genome there are 23 pairs of strands of DNA which we call chromosomes. And the strands are wrapped around protein complexes called nucleosomes, which consist of proteins called histones.
3. DNA methylation and epigenetics
DNA methylation is one of three central mechanisms for epigenetic regulation of gene expression. My skin, toes, heart, liver and brain all have the same genes. What makes them so different? It is epigenetic (beyond genetic) expression during fetal development. At 86 now, I look quite different than I did at 30 or 3, but yet I have the same genes I was born with. Why do I seem to be so different now? It is because of epigenetically-driven changes in gene expression in the course of aging from conception to now.
Our protein-encoding genes are mostly with us for life but other components of our DNA are highly dynamic. Specifically, DNA embodies elements and processes that regulate gene expression. We call these regulatory elements and processes epigenetic ones. They are different in different cell types and organs and evolve with aging. DNA methylation is one of the important epigenetic mechanisms, the subject of this blog entry. Methylation patterns in our genome can be profoundly impacted by environmental factors from conception to death. There is very strong evidence they can be changed or modified by our lifestyle, our diet, our illnesses, and in some cases even by the lifestyle and diet and illnesses that our parents and grandparents had. These methylation patterns are major drivers of gene expression that change with aging and exercise profound effects on our health and longevity. Methylation molecules can bind to a gene to get it either ignored or enhanced and so, turn it on or off. If the promoters of protective genes like P53 become methylated in the gene’s promoter “CpG islands,” the gene cannot be activated to produce its protein. Cancer or several other pathologies can result. If the non-CpG islands of numerous normally methylated and inactive genes become gradually demethylated with aging, all kinds of unwanted gene expression can occur leading to numerous pathologies and accelerated aging. This blog entry gets into detail about these matters.
DNA cytosine methylation is only one of the three main “pillars” of epigenetic regulation of gene expression. The other two are histone tail modifications and microRNA regulation of mRNA. We have previously written about histone tail modification in several of our blogs. See for example (ref), (ref), (ref), and (ref). A few items in this current blog entry are also concerned with these other two pillars.
- DNMT methyl transferase enzymes and TET enzymes
Cytosine methylation is a heritable feature of DNA that can be passed down from generation to generation, maintaining the same DNA methylation patterns in offspring. The enzymes that recognize this “Cytosine-phosphate-Guanine” dinucleotide motif are called DNA methyltransferases (DNMTs) and transfer the methyl group from the methionine cycle molecule called “S-adenosyl methionine”, or SAM. There are a number of these including DNMT1, DNMT2, DNMT3A, DNMT3B and DNMT3L. DNMT enzymes are specialized into ones used to replicate the DNA methylation pattern as DNA is replicated during the S-phase of mitosis and meiosis (DNMT1) and enzymes that do de novo methylation of CpGs that were not methylated in the original DNA (DNMT3a & DNMT3b). There is a considerable body of literature associated with such enzymes, and selective inhibition of some of them has been proposed as an anti-cancer therapy. “The levels of DNMTs, especially those of DNMT3B, DNMT3A, and DNMT3L, are often increased in various cancer tissues and cell lines, which may partially account for the hypermethylation of promoter CpG-rich regions of tumor suppressor genes in a variety of malignancies(ref).”
There are also enzymes that can remove this methyl group from the cytosine at these CpG residues, called “ten-eleven transferase”, or TET. Using oxygen as an oxidizer, TET enzymes oxidize the methyl-cytosine to hydroxy-methylcytosine, then to formylcytosine, and then to carboxymethylcytosine via the hydroxlase activity of the TET enzymes. Another enzyme called TDG then removes the carboxyl group, restoring the cytosine to its original unmethylated state.
- About gene promoter regions, CpG islands, introns, 5’-UTRs, 3’-UTRs, exons, and retrotransposons
These are matters referred to in the more-technical parts of this blog entry. Please don’t be scared off by them if you don’t know any genetics. If you are seriously to follow longevity or other key areas of life sciences research in the future you will come across them again and again. Here is a little crib sheet to help you read and understand what follows.
Gene and Gene promoter and terminator regions
A gene is an identifiable locatable functional unit within a strand of DNA that encodes information, such as for a protein. A gene has a beginning known as a promoter area and an end known as the terminator, encompassing a specific region in the DNA. A given gene is always located in the same position in different organisms of the same species. For different humans, for example, the chromosome number 6 always contains the same genes in the same order. The same is true for mice, moose, mongoose and marsupials. There are an estimated 19,000 to 20,000 protein-coding genes in the human genome.
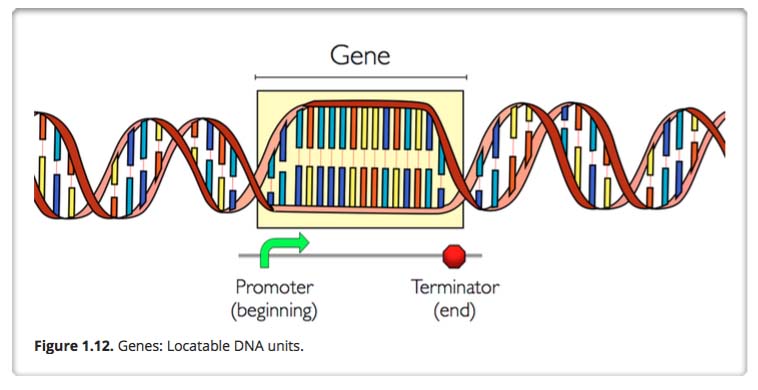
Despite what you may have imagined, only about 1.5% of DNA codes for genes. The rest of the DNA seems to fulfill regulatory functions of various kinds, though the workings of these are not yet fully understood. About half of the remaining 98.5 percent of DNA is made up of transposable elements, or DNA that can copy itself within the genome much like a virus can. Increasingly. we are seeing how these transposable entries too play important roles in gene regulation and triggering evolution. You might want to review our recent blog elements on transposable DNA elements: Part 1: basics and importance, Part 2: The Self-copy Machines in Your Genes, and Part 3: TEs and and other key mechanisms of evolution: incRNAs, A to I editing, alternative splicing and exonization.
Introns, exons, codons, open reading frames
Introns and exons are parts of genes. Exons code for proteins; during gene transcription they are what is converted to messenger RNA, a step toward making the protein encoded by the gene, Introns do not code for proteins and are skipped over during splicing after transcription, at least in the case of normal splicing. There are an average of 8.8 exons and 7.8 introns per human gene. A codon is a triplet of adjacent nucleotides in the messenger RNA chain that codes for a specific amino acid in the synthesis of a protein molecule(ref)” Start and stop codons are nucleotide triplets within messenger RNA that signal initiation and termination of translation into proteins by a ribosome.. An Open Reading Frame (ORF) is a region of the nucleotide sequences from the start codon (ATG) to the stop codon.
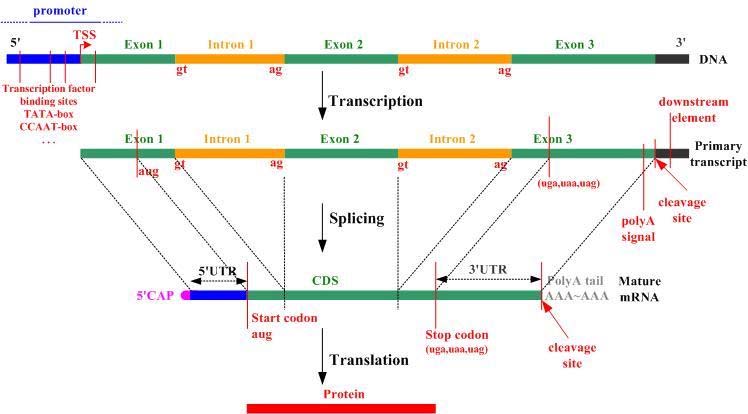
Untranslated regions: 5′ UTR and 3′ UTR
These are regions of DNA at the head and tail of a gene as shown in the above diagram. A 5′ UTR often precedes a start codon.
CpG island
Key to understanding DNA methylation is learning what a CpG island is (abbreviated as “CGI”). The majority of CpG dinucleotides outside of the promoter region in the human genome are normally methylated. But within certain regions often associated with promoters, 5’-UTRs, introns, and to a lesser extent, 3’-UTRs and retrotransposons; there are CpG rich regions called “CpG islands” (aka CGIs) where gene regulation occurs by the process of cytosine methylation within the CpG island cytosines. This type of gene regulation is one of the three major kinds of epigenetic regulation. A CGI is a sequence of “Cytosine-phosphate-Guanine” DNA dinucleotide repeats that are > 200 base pairs long and where at least 55% of DNA bases in the sequence are CpGs. Furthermore, to be a CpG island (CGI), the sequence must have an observed-to-expected ratio of > 65% CpGs. (The old definition was 50% CpGs and 60% observed-to-expected ratio, but this was changed in 2002). Not all genes have CGIs, but in mammals, 70% of protein coding genes have CGIs in their promoters. The genes that have CGIs in their promoter regions include “house keeping” genes (which are constitutively expressed) as well as some regulated genes that are only expressed at certain times. In addition to having a CGI in the promoter, there are two other features of mammalian promoters: 1) low nucleosome occupancy (no histone “spool” winding at these promoters), and the 2) pre-association of RNA Pol II at the promoter site in inactive genes.
70% of mammalian genes contain a CGI in their gene promoters. However, only 35% of all CGIs are found in the promoter region of genes. The other 65% of CGIs are found within repetitive elements (10%), introns (7.7%), the “head” of a gene (5′-UTR) and the “tail” of a gene (3′-UTR). CGIs in promoters are mostly unmethylated, whereas CGIs in introns are differentially methylated, changing their methylation state in a dynamic fashion, in response to various external stimuli, such as cortisol-glucocorticoid receptor binding.
Here is an alternative definition fof CGIs rom Wikipedia:
“The CpG sites or CG sites are regions of DNAwhere a cytosine nucleotide is followed by a guaninenucleotide in the linear sequence of bases along its 5′ → 3′ direction. CpG is shorthand for 5’—C—phosphate—G—3′ , that is, cytosine and guanine separated by only one phosphate; phosphate links any two nucleosides together in DNA. The CpGnotation is used to distinguish this single-stranded linear sequence from the CG base-pairing of cytosine and guanine for double-stranded sequences. The CpG notation is therefore to be interpreted as the cytosine being 5 prime to the guanine base. CpGshould not be confused with GpC, the latter meaning that a guanine is followed by a cytosine in the 5′ → 3′ direction of a single-stranded sequence.”
SECTION B – DNA METHYLATION AND AGING
– CLOCKS BASED ON DNA METHYLATION MEASURES ARE THE BEST CLOCKS FOR AGING (though these clocks are not the only ones).
The remainder of this blog entry consists mainly of Jim Watson writings, with some substantive contributions as well as editorial work by Vince Giuliano
The items below describe several facets of the story.
The two faces of DNA methylation dysregulation
Again as basic, there are two faces to age-related changes in DNA methylation; 1. GpC island promoter sites where progressive hypermethylation occurs with aging, and 2. A gradual loss of DNA methylation throughout the entire genome. This occurs in many regions of the genome and is observed in a variety of species including humans. This is, often referred to as global DNA hypomethylation or epigenetic drift – a gradual loss of DNA methylation in CpGs found in promoters, exons, introns, gene bodies, intergenic regions, and interspersed repetitive sequences (transposable elements), including human endogenous retroviruses (HERV), Alu repeats, and other repetitive DNA. These same two kinds of methylation changes occur in cancers and certain other disease processes. Later we discuss what are known to cause them and their implications for aging and diseases. We discuss these two faces in Section C of this blog entry.
1. DNA methylation is the best biomarker for aging
The development of “DNA methylation biomarkers of aging” is a new and exciting field that may also lead us to discover some of the fundamental causes of aging. Changes in DNA methylation correlates with aging, as do shortening telomere lengths – though both approaches give only approximate age estimates. DNA methylation provides the best clocks for aging known, far more reliable than telomere length. We start with Steve Horvath’s 2013 publication about “DNA methylation clock” that accurately predicts chronological age (3X more accurately than telomere length) has 353 methylation sites in the human genome where methylation changes as a function of aging. This 353 site “clock “can accurately predict how old a cell is or how old an organ is to within 2 years, chronologically. Slightly less than 1/2 of these 353 sites are CpG sites where methylation INCREASES. As you would expect, these are mostly in gene promoter “CpG islands” and this means that the gene expression of these genes are down-regulated (Ex: gene for FoxN1). Slightly more than 1/2 of the 353 CpG sites are places where methylation DECREASES. As you would expect, these sites are NOT in gene promoter CpG islands. The result is increasing activation of many genes including ones we would prefer to remain silent with aging.
Reference: 2013 DNA methylation age of human tissues and cell types
“It is not yet known whether DNA methylation levels can be used to accurately predict age across a broad spectrum of human tissues and cell types, nor whether the resulting age prediction is a biologically meaningful measure. Results: I developed a multi-tissue predictor of age that allows one to estimate the DNA methylation age of most tissues and cell types. The predictor, which is freely available, was developed using 8,000 samples from 82 Illumina DNA methylation array datasets, encompassing 51 healthy tissues and cell types. I found that DNA methylation age has the following properties: first, it is close to zero for embryonic and induced pluripotent stem cells; second, it correlates with cell passage number; third, it gives rise to a highly heritable measure of age acceleration; and, fourth, it is applicable to chimpanzee tissues. Analysis of 6,000 cancer samples from 32 datasets showed that all of the considered 20 cancer types exhibit significant age acceleration, with an average of 36 years. Low age-acceleration of cancer tissue is associated with a high number of somatic mutations andTP53 mutations, while mutations in steroid receptors greatly accelerate DNA methylation age in breast cancer. Finally, I characterize the 353 CpG sites that together form an aging clock in terms of chromatin states and tissue variance. Conclusions: I propose that DNA methylation age measures the cumulative effect of an epigenetic maintenance system. This novel epigenetic clock can be used to address a host of questions in developmental biology, cancer and aging research.”
In 2015 Horvath and colleagues published another paper concluding that DNA methylation-derived measures of accelerated aging are heritable traits that predict mortality.
Reference: 2015 DNA methylation age of blood predicts all-cause mortality in later life
“DNA methylation levels change with age. Recent studies have identified biomarkers of chronological age based on DNA methylation levels. It is not yet known whether DNA methylation age captures aspects of biological age. Results: Here we test whether differences between people’s chronological ages and estimated ages, DNA methylation age, predict all-cause mortality in later life. The difference between DNA methylation age and chronological age (Δage) was calculated in four longitudinal cohorts of older people. Meta-analysis of proportional hazards models from the four cohorts was used to determine the association between Δage and mortality. A 5-year higher Δage is associated with a 21% higher mortality risk, adjusting for age and sex. After further adjustments for childhood IQ, education, social class, hypertension, diabetes, cardiovascular disease, and APOE e4 status, there is a 16% increased mortality risk for those with a 5-year higher Δage. A pedigree-based heritability analysis of Δage was conducted in a separate cohort. The heritability of Δage was 0.43. Conclusions: DNA methylation-derived measures of accelerated aging are heritable traits that predict mortality.”
2. Telomere length and Epigenetic clocks are independently associated with aging and mortality.
But, even together the two clocks explain only a small part of the variance encountered in aging.
This is the basic finding of a landmark paper that just came out in March, 2016. They found that telomere length decreased at a rate of 48-67 base pairs per year, and that telomere length only explained 6.6% of the variance in age. The correlation of telomere length with aging is about 0.30. They found that DNAm epigenetic clock age explained 10% of the variance in age but had a high correlation with aging = 0.95. When both telomere length and DNAm epigenetic age were combined, they explained only 17.3% of the variance in age. Thus the two factors independently predict aging.
One standard deviation increase in epigenetic age increased mortality by 22%. One standard deviation increase in baseline telomere length decreased mortality by 11%. Interestingly, both telomere length and epigenetic age did not correlate with chronological age in the 70-90 year olds. This suggests that in old age, other factors are more important than telomere length and DNAm age.
References:
2013 A systematic review of leukocyte telomere length and age in adults
3. At least five epigenetic DNA methylation clocks have been described
Here they are, two by Horvath and three others:
110 CpG clock –
Horvath described this DNAm clock in 2013, in the same paper as the 353 CpG clock (it is a subset of the 353 CpGs) Cor = 0.95 for training data and test data. Like the 353 CpG clock, this one was based on 21,369 CpG sites on an Illumina chip.
353 CpG clock –
Horvath described this one, sifting through 21,369 CpG sites Cor = 0.97 in training data, 0.96 in test data, with an error bar of + 3.6 yrs. 193 sites were hypermethylated with aging, which were mostly in Polycomb-Group protein binding sites. 160 of the 353 CpG sites were hypomethyated and CpG shores were over-represented in this group. Other researchers have noted that 85 of the 353 CpG sites involved glucocorticoid responsive genes
3 CpG clock –
Weidner described a very simple methylation clock that only has 3 CpGs (2014)! Cor = 0.85. This is quite amazing! Estimates age + 4.5 yrs on training set and 5.6 yrs on the validation set. CpG sites are on the genes (ITGA2B, ASPA, and PDE4C). There is a German company that will do commercial testing on samples for research purposes that uses this 3 CpG clock.
73 CpG clock –
Hannum described this DNAm clock in 2013, from only whole blood. Cor = 0.96, with + 3.6yrs. This CpG DNA clock was based on the 485,577 CpG Illumina chip. 70,387 of the 485K CpG sites (15%) correlated with aging. 44% of these 70,387 sites increased in methylation with aging whereas 56% of these 70,387 sites underwent age-associated decrease in methylation.
102 CpG clock –
Weidner also described this DNAm clock in the same 2014 paper as the 3 CpG clock paper. This was used to find/select the 3 CpG sites and could accurately predict chronological age within 3.34 years. Like the Horvath and Hannum DNAm clocks, this one is a mixture of both hypomethyated (58) & hypermethylated (44) CpG sites. More details on these residues below.
References:
2013 Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates (Hannum et al.)
2014 Aging of blood can be tracked by DNA methylation changes at just three CpG sites (Weidner et al.)
4. Details and Differences in Epigenetic Clocks
Weidner’s 3 CpG clock and their 102 CpG clock both claim to correlate with telomere length. Hunnum did not make that claim and in 2016, another group showed that the Hannum CpG clock independently predicted age and mortality, compared to telomere length (see Item 3 above on this).
Hannum’s 71 CpG clock showed that men undergo faster epigenetic aging than women by 4%. Horvath’s 353 CpG epigenetic clock showed that the female breast aged faster than the rest of the female body by 7-8 yrs. The methylation marker cg27193080 correlated the best with aging in Hanum’s DNAm clock and napped to the gene for Methyl binding protein 4 (MBP4), which is a Methyl binding protein involving DNA repair. Seven other validated specific differentially methylated sites with aging were on the following genes: NEK4, JAKMIP3, GTPBP10, and STEAP2 in Hanum’s DNAm clock. Although a list of genes is available of the CoG residues in Horvath’s clock, he did not mention any specific genes associated with DNAm sites, but mentioned that many of the hypermethylated CpG sites were in regions with bivalent chromatin domains (I.e. Silent, but poised for gene transcription) which is often seen with stem cell genes.
Interestingly, Weidner also showed that in their 102 CpG clock, many of the CpG residues that became hypomethyated with aging (44 residues) were hypermethylated in both embryonic stem cells and in iPSC cells, suggesting that these CpG sites we’re in stem-cell related genes that were reprogrammed with iPSC development.
References:
2013 Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates (Hannum et al.)
2014 Aging of blood can be tracked by DNA methylation changes at just three CpG sites (Weidner et al.)
5. Primate Epigenetic Aging is an Interspecies phenomena demonstrating yet-again that aging is programmed
Chimpanzees, Bonobos, and humans share the same epigenetic clock, as shown by Horvath’s elastic net regression algorithm that agnostically chose 353 CpGs, whereas Gotillas did not share this clock (too distant from humans in evolution). This goes against environmental factors and extrinsic factors as the “cause” of aging, and suggests that the pattern of aging may be programmed and heritable across different species (I.e. It is genus/family-specific type of programmed aging).
6. Cell passage number correlates with DNA methylation age.
This also supports the suggestion that normal cell mitotic activity regulates lifespan. This hypothesis is supported by Weidner’s conclusion, but not by the new 2016 article that used Hanum’s DNAm clock.
7. Epigenetics including DNA methylation plays a major role in the pathogenesis of Aging
Recently, it has become clear that epigenetic plays a major role in aging. As with cancer, many genes undergo CGI hypermethylation, resulting in gene silencing, even with no signs of DNA mutation. Examples of this include the Helicase gene and lamin A/C genes. Whereas in Werner’s syndrome (an accelerated aging disease), the helicase gene is mutated, with aging the Helicase gene in aging is silenced by DNA methylation of the CGI in its promoter region. Whereas in Hutchinson-Gilford-Progeria syndrome (another accelerated aging disease), the lamin A/C gene is mutated, with aging, the cryptic splice site in the lamin A/C gene is “turned on” by epigenetic mechanisms that are still unclear. Thus epigenetics explains many of the classic features of normal aging that are seen in accelerated aging in progeroid disease syndromes.
Many other “more-ordinary” genes undergo CpG island hypermethylation with aging, including genes for tumor suppression (COX7A1, LOX, RUNX3, TIG1, p16INK4a, RASSF1, DUSP22), development and growth genes (IGF2, cFos), cell-cell adhesion (CDH1), metabolism (ELOVL2, SLC38A4, SLC22A18, MGC3207, ECRG4, ATP13A4, AGPAT2, and LEP), DNA repair (MLH1), and control of signal transmission (FZD1, FZD7). All of these genes exhibit altered gene expression with aging.
The expressión of the DNMT genes is also impaired with aging. There is an under-expression of DNMT1 and DNMT3A in lymphocytes from elderly people. There is an increase in expression of DNMT3B with aging. Thus these genes may play a major role in differential DNA methylation as a function of aging.
Reference: 2012 Epigenetics and aging
Another epigenetic feature of aging is the gradual loss of X-chromosome silencing of the inactive X chromosome in females, especially in the myeloid cell lineages of peripheral blood cells. A correlation between the loss of X-chromsome gene silencing of the inactive X chromosome is also seen in cancer, autoimmune diseases, and other diseases.
As mentioned previously, a central epigenetic feature of aging is the gradual loss of DNA methylation throughout the entire genome. This occurs in many regions of the genome. There is a gradual loss of DNA methylation in CpGs found in promoters, exons, introns, gene bodies, intergenic regions, and interspersed repetitive sequences (transposable elements), including human endogenous retroviruses (HERV), Alu repeats, and other repetitive DNA.
Reference: 2012 Epigenetics and aging
Another epigenetic mechanism of aging involves the acetylation/deacetylation of specific lysine on histone proteins. One example of this form of epigenetic aging involves the loss of normal deacetylation of lysine 16 on histone H4 (H4K16), which is due to the loss of Sirtuin enzyme function. The exact cause of SIRT1 function loss with aging is still unclear, but appears to be linked to the age-associated decline in NAD+ levels and the decline in the NAD/NADH ratio within cells, especially the nucleus of the cell. Discussion of this can be found in our blog entries on the NAD world (ref), (ref), (ref), (ref), (ref). In particular, see 30 Major Factors that Control SIRT1 Expression, SIRT1 Activity, and SIRT1-mediated Aging. Part 3 of the series NAD+ an emerging framework for health and life extension.
Here is an illustration of the role of epigenetics in both cancer and in aging:
References:
2007 Epigenetics and aging: the targets and the marks
2012 Epigenetics and aging
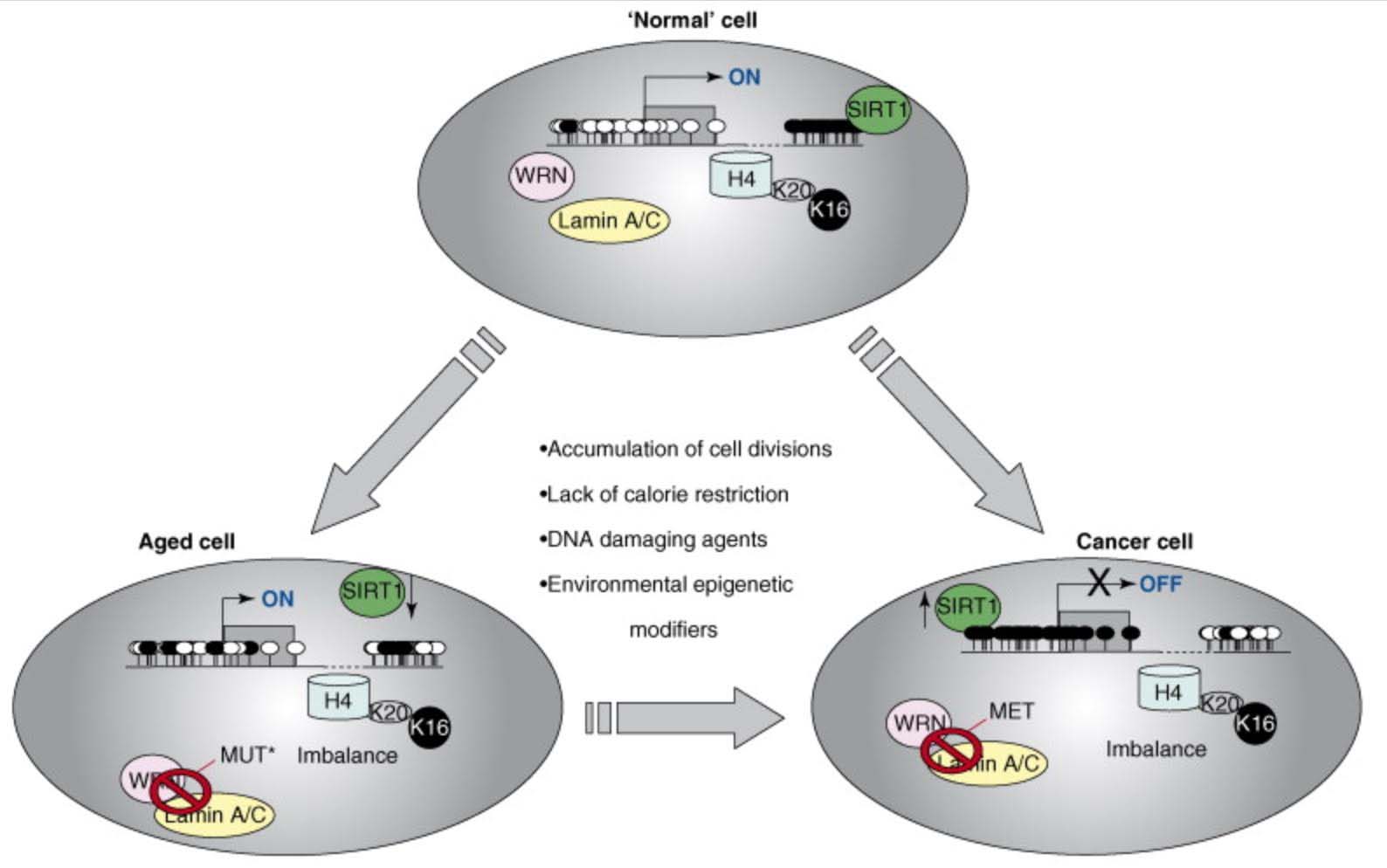
This diagram provides a very strong endorsement of the use of DNA methylation as an accurate “clock,” Additional reference: 2016 Quantitative comparison of DNA methylation assays for biomarker development and clinical applications
8. Major epigenetic changes that happen with aging
As I mentioned above, several generalized changes in epigenetics occur with aging and several specific changes in epigenetics occur with aging. Here are the changes in a short list:
a) Epigenetic drift (aka global hypomethylation of CpGs in the genome) – 15%
With normal aging, Epigenetic drift produces specific (not stochastic) distinct hypomethylation of satellite DNA, transposable elements (LINEs, SINEs, HERV LTRs), gene bodies, and intergenic regions of DNA. The term “Epigenetic drift” does not describe the hypermethylation of CGIs that also occurs with aging. It only describes the hypomethylation that occurs with aging. Over a lifetime, the total quantitative “drift” amounts to approximately an 11-15% change in DNA methylation of the entire genome (Ref: Hannum et. al., 2013). The pattern of differential DNA methylation with aging is distinct and not stochastic, and is often referred to as the “aging methylome”. This global hypomethylation feature of aging was used to make up 160 of the 353 CpG sites in Steve Horvath’s “DNA methylation clock” that were found to correlate closely with chronological age, using a computer algorithm with an elastic net regression model. (See Section B above.) Interestingly, the hypomethylated sites chosen by computer algorithm included an over-representation of hypomethylated cytosines in GpG shores (the borders of CpG islands), not gene bodies, transposable elements, or satellite repeats. More recently, a specific histone marker for these regions of global hypomethylation has been identified by Fernandez and colleagues – the H3K4me1 mark. With global hypomethylation, the specific sites that are hypomethylated develop the H3K4me1 mark. Thus, specific histone changes correlate with specific hypermethylation of DNA that are associated with global DNA aging.
References:
2013 DNA methylation age of human tissues and cell types
2015 H3K4me1 marks DNA regions hypomethylated during aging in human stem and differentiated cells
b) Locus-specific Hypermethylation of CpGs with aging (in and outside promoter CGIs)
With normal aging, many CpG residues become hypermethylated and the sites undergoing hypermethylation are specific sites, not random sites (aka “locus specific hypermethylation”). Although conventional wisdom was that these hypermethylated CpGs were all found within promoter CpG islands, this generalization may not be accurate. At the least, this view is an oversimplification of the true phenomena of age-related hypermethylation sites. For instance, in Steve Horvath’s computer algorithm model, 193 of the 353 CpG residues chosen by computer were hypermethylated CpGs. Although CpG residues in CGIs were included in the 193 sites, the methylcytosines chosen by the unbiased computer program did not show any over-representation of CpG residues found in CpG islands, though most other authors have found that hypermethylated CpGs were mostly in CpG islands.
References:
2013 DNA methylation age of human tissues and cell types
c) Loss of X inactivation with aging
With aging, there is a gradual loss of silencing (imprinting) of the 2nd X chromosome in females. This phenomenon is seen both with normal aging as well as in accelerated aging syndromes such as HGPS and Werner’s syndrome. Both changes in histone tails (H3K27me3) is lost and DNA methylation of CpGs is lost on the inactivated X chromosome, which allows both genes and transposable elements on the X chromosome to be transcribed. The loss of DNA methylation can be easily measured on the X chromosome in peripheral white blood cell DNA and could be used as part of a “DNA methylation clock”, although none of Horvath’s 353 CpG sites were found on the X chromosome.
d) Differential DNMT expression with aging
Another consistent finding in aged cells is the up-regulation of DNMT3b and the down regulation of DNMT1 and DNMT3a metyltransferase enzymes. Whereas the decrease in gene expression of DNMT1 and DNMT3a were due to hypermethylation of their CpG islands (CGIs), the increase in DNMT3b was not due to methylation in a promoter CpG, but was due instead to CpG methylation of a residue outside of the CpG island (p352). Since DNMT1 is the isoform that methylates the newly synthesized strand of DNA during DNA replication (S phase of cell cycle), this means that dividing cells loose some of the normal pattern of DNA methylation with aging, due to the down-regulated DNMT1. The differential expression of DNMT3a (decreased) and DNMT3b (increased) is thought to be one of the main reasons for the age-specific, locus-specific pattern of differential DNA methylation with aging.
References:
AWSAccessKeyId=AKIAJ56TQJRTWSMTNPEA&Expires=1470050452&Signature=CrnwsfPnVm9w6jCcyukbuvJxpUw%3D&response-
e) Specific genes undergo CpG hypermethylation with aging, some in CGIs and some outside of CGIs
Another consistent feature of aging includes the epigenetic silencing of specific genes by CpG hypermethylation. Some of these are within CpG islands (CGIs) and some are outside of CGIs. For instance, the following genes are commonly silenced with aging due to CGI hypermethylation, including ESR1, GSTP1, IGF2, MGMT, MYOD1, RARB, RASSF1, COX7A1, LOX RUNX3, TIG1, p16INK4a, RASSF1, DUSP22, IGF2, cFos, CDH1, ELOVL2, SLC38A4 SLC22A18, MGC3207, ECRG4, ATP13A4, AGPAT2, LEP, MLH1, FZD1, and FZd7).
Although there are fewer genes that undergo age-specific DNA hypermethylation of non-CpG island sites, there are several that do change with aging, including DNMT3B, which undergoes CpG methylation outside of the CGI and whose expression increases with aging; HDAC7A which undergoes CpG methylation outside of a CGI. Exactly why these genes undergo CGI hypermethylation and not others is not clear.
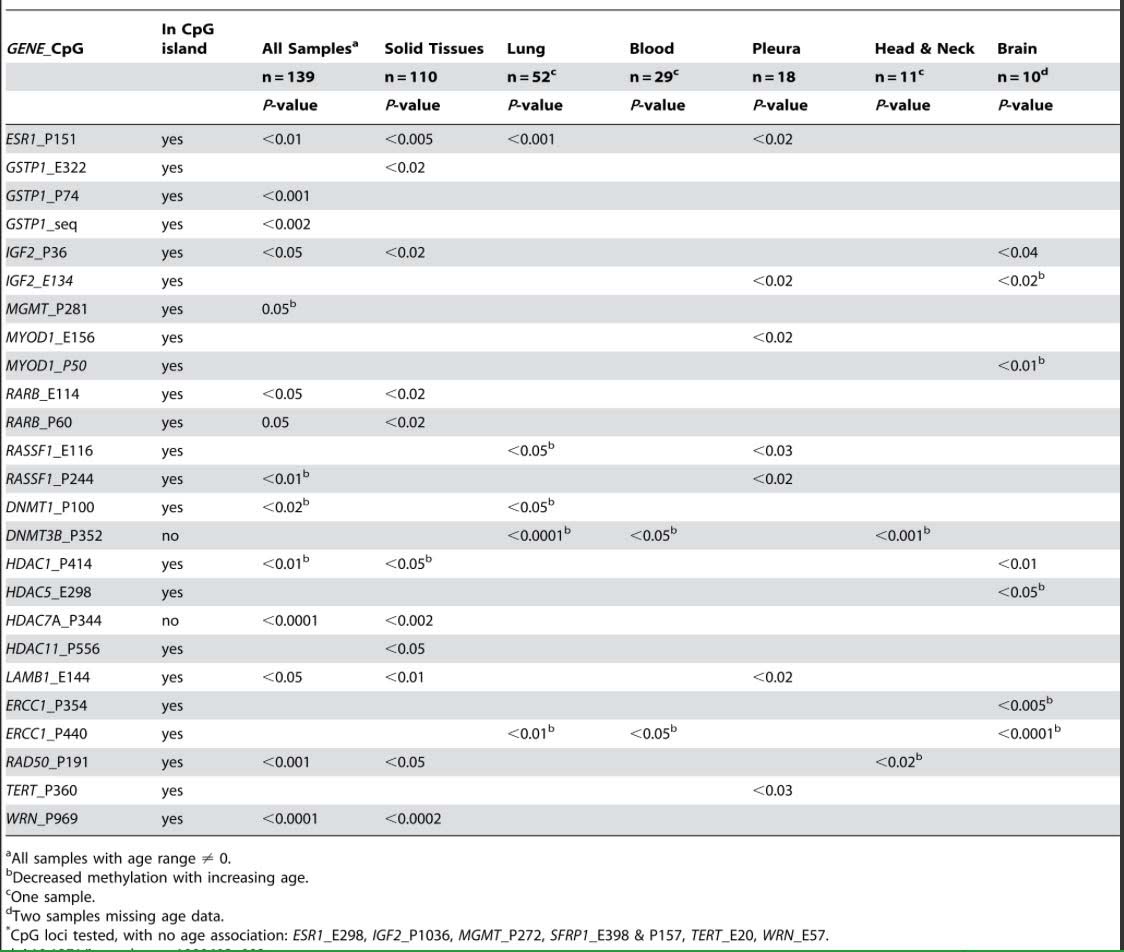
Reference and table source: 2009 Aging and Environmental Exposures Alter Tissue-Specific DNA Methylation Dependent upon CpG Island Context
f) HGPS is an “accelerated epigenetic aging” phenomena.
Hutchinson-Gilford Progeria is due to de novo mutations in the LMNA gene, most commonly the “C1824T mutation” With C1824T, the spliceosome uses a cryptic splice site found in exon 11 of the LMNA gene, resulting in the production of a “mutant lamin A protein” called “progerin” that is missing 50 amino acids in the middle of the protein. With normal aging, this same cryptic splice site is also activated, but at a rate that is 50-fold lower. However, UVA light exposure can increase the use of this cryptic splice site by alternative splicing, accelerating skin aging in those that tan or sunbathe (UVA is the light used in tanning booths). Regardless of whether progeria is produced by the C1824T mutation or by normal aging or by UV-A light, the accelerated aging is primarily mediated by epigenetics. Specifically, the histone mark of normal heterochromatin (H3K27me3) is lost on the inactivated X chromosome, resulting in transcription from the X chromosome. The histone mark of heterochromatin that is found near all of the centromeres (H3K9me3) is also lost, as is the heterochromatin protein 1a (Hp1a) which is also associated with pericentric heterochromatin. As a result of the loss of pericentric heterochromatin H3K9me3 and Hp1a, there is an up-regulation in the transcription of pericentric satellite III repeats. In the histone 4 subunit, the opposite occurs – there is an increase in H4K20 trimethylation (H4K20me3). See our blog entry Hutchinson-Gilford Progeria Syndrome – a disease of accelerated aging due to Alternative Splicing.
References:
2006 Lamin A-Dependent Nuclear Defects in Human Aging
2014 Epigenetic involvement in Hutchinson-Gilford progeria syndrome: a mini-review
2013 Longwave UV Light Induces the Aging-Associated Progerin
2006 Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging
g) The Werner’s syndrome gene undergoes CpG hypermethylation
Unlike HGPS, where alternative splicing produces a mutant protein, the problem in Werner’s syndrome is a mutation in the gene for a DNA unwinding gene called “Werner’s helicase”, or WRN. In normal aging, this gene is not mutated, but is consistently silenced by CpG methylation. Thus normal aging is accompanied by many of the same features of Werner’s syndrome, due to a loss of gene expression of WRN.
Reference:2009 Aging and Environmental Exposures Alter Tissue-Specific DNA Methylation Dependent upon CpG Island Context
9. Histone based “epigenetic aging” phenomena – ↓H3K9me3, ↓H3K27me3, ↑H4K16Ac, ↑H4K20me3, ↑H3K4me1
Although there are many changes to histone tails that occur with aging, there are 5 specific changes in histone modifications that occur on the lysine (K) side changes that appear to be very consistent with aging. They include declines in H3K9 trimethylation (H3K9me3) and H3K27 trimethylation (H3K27me3), which are considered repressive histone marks; as well as increases in H4K16 acetylation (H4K16Ac), H4K20 trimethylation (H3k20me3), and H3K4 methylation, which are associated with increased gene transcription. Of these 5 consistent changes in histone modifications, the one that is linked closely to SIRT1 function is H4K20. Here is a diagram on this:
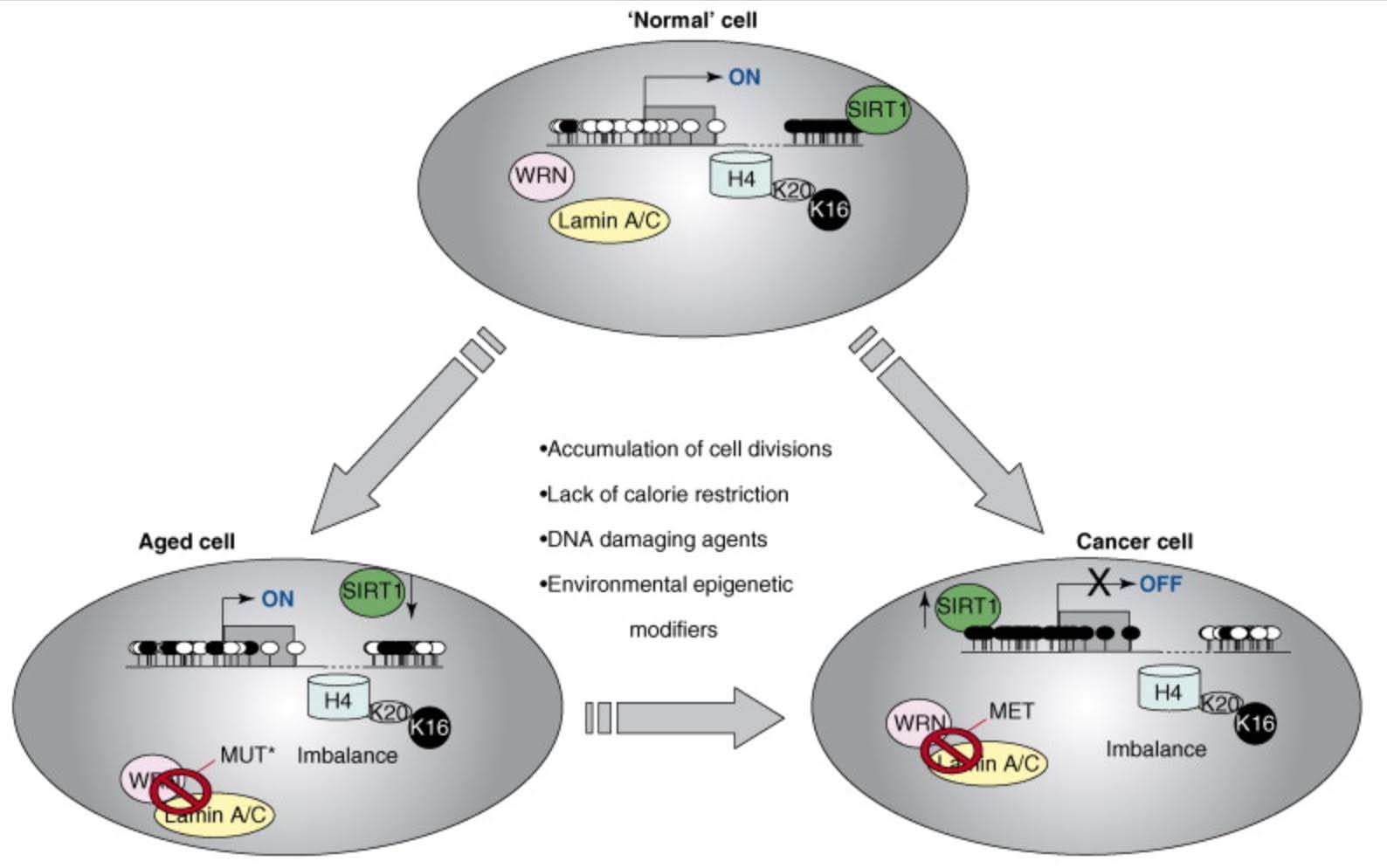
Reference and image source: 2015 H3K4me1 marks DNA regions hypomethylated during aging in human stem and differentiated cells
10. Age, Cancer and Inflammation-associated hypermethylation occurs at Polycom Group Protein target genes and may be the “quasi-program of aging”
(Please read this item, read #11, below and then come back and re-read this item – they are both related, but are not exactly talking about the same genes)
What Polycomb Repressor Complexes (PcGs) do
In 2010, Rakyan and colleagues showed that age-associated hypermethylated DNA sites frequently occurred in promoters with bivalent chromatin domains (see #11 below). This terminology described genes where there was histone modifications that showed both repressive (off) and active (on) states. This effect was often seen in neural stem cells. The hypothesis was that these genes were “poised for action.” Since 2010, it has become increasingly clear that a more important feature of these age-associated DNA hypermethylated genes was that they were Polycomb group protein (PcG) target genes. In 2015, there was enough evidence to show that this PcG target gene “hypermethylation pattern” occurred in both aging and cancer. Polycomb group proteins form complexes that associated with DNA and chromatin and normally function as repressors of genes involved in embryonic development and cell fate decisions.
There are two Polycomb group protein repressor complexes referred to as PRC1 and PRC2. The function of PRC2 is to tri-methylate lysine 27 on histone subunit H3 (creating H3K27me3). This silences the gene which subsequently (or independently) becomes bound by PRC1 protein complexes, which leads to further chromatin compaction and stabilizes the silencing of the gene.
The link between PcGs and gene hypermethylation
Polycomb group protein target genes have been shown to be prone to hypermethylation in many different types of cancers and these same genes have been shown to be hypermethylated with aging. Why is cancer and aging associated with PcG target genes? This question has not been conclusively answered, but one theory is that DNA methylation patterns are involved in regulating PcG accessibility and vice versa (i.e. PcG accessibility regulates DNA methylation). What is clear is that a high density of unmethylated CpG dinucleotides is sufficient for PcG recruitment. Also H3K27me3 histone occupancy (aka appearance or recruitment) occurred at regions of the genome that are normally highly DNA-methylated and then become demethylated. In other words, Polycomb complexes recognize unmethylated DNA regions. This occurs for the protein KDM2B. KDM2B recruits PRC1 and PRC2.
Specific Polycomb Group protein target genes that are hypermethylated in aging and cancer
Specific genes where hypermethylation occurs in both cancer and aging include IGF2, hypermethylated in cancer 1 (HIC1), caspace-8 (CASP8),glutathione S-transferase 1 (GSTP1), suppressor of cytokine signaling (SOCS1), RAS association domain family 1A (FASSF1A), p16/CDKN2A, adenomatosis polyposis coli (APC), and the estrogen receptor 1 (ESR1). DNA hypermethylation of these genes occurs in both aging and several types of cancers. Also DNA hypermethylation in HOX genes or in certain genes encoding lineage-specific transcription factors in non-small cell lung cancer could represent an epigenetic component of an age-related process.
Specific Polycomb Group protein target genes that are hypermethylated in inflammatory diseases
A similar association between PcG target genes and DNA hypermethylation has been seen in inflammatory diseases. This occurs in ulcerative colitis, Crohn’s disease, Rheumatoid arthritis, Systemic Lupus Erythematosis, and other diseases. In these diseases, DNA hypermethylation occurs in genes recognized by Polycomb complexes, PRC1 and PRC2. However, not all genes undergo hypermethylation in inflammatory diseases. For instance, the TNF-alpha gene shows a decrease in promoter hypermethylation in inflammatory diseases, resulting in an increase in transcription of the TNF-alpha gene. Also in atherosclerosis, promoter CpG hypomethylation also occurs. The same feature is seen in Alzheimer’s disease. Thus the hypermethylation of genes does not occur in all genes – .only those associated with Polycomb protein complexes.
Methylation of Polycomb Target Genes is Mediated by Inflammation
The mystery of why Polycomb protein Complex target genes become hypermethylated in both aging and cancer is finally starting to become clear. The link is inflammation. As mentioned above, PcG target gene hypermethylation of DNA was seen in inflammation. In several different research labs, it has been independently confirmed that inflammation is the “driver” of the DNA methylation in the Polycomb Target Genes. (Hahn, et.al, Cancer Research, 2008). Chronic inflammation is thought to causally contribute to as much as 25% of all cancers worldwide. In a mouse model of inflammatory bowel disease (IBD), it was shown that inflammation precedes DNA hypermethylation of PcG target genes with 70% of the 250 genes noted to be hypermethylated in IBD being stem cell genes and 59% of the hypermethylated genes being marked with H3K27me3 histone marks. Acquisition of DNA methylation at CpG islands was associated with the loss of H3K27me3 pattern and this occurred only in the cells with inflammation and confirmed age-dependent differential methylation. Here are some Venn diagrams that show the overlap between aging and inflammation.
Reference: 2015 Aging and DNA methylation
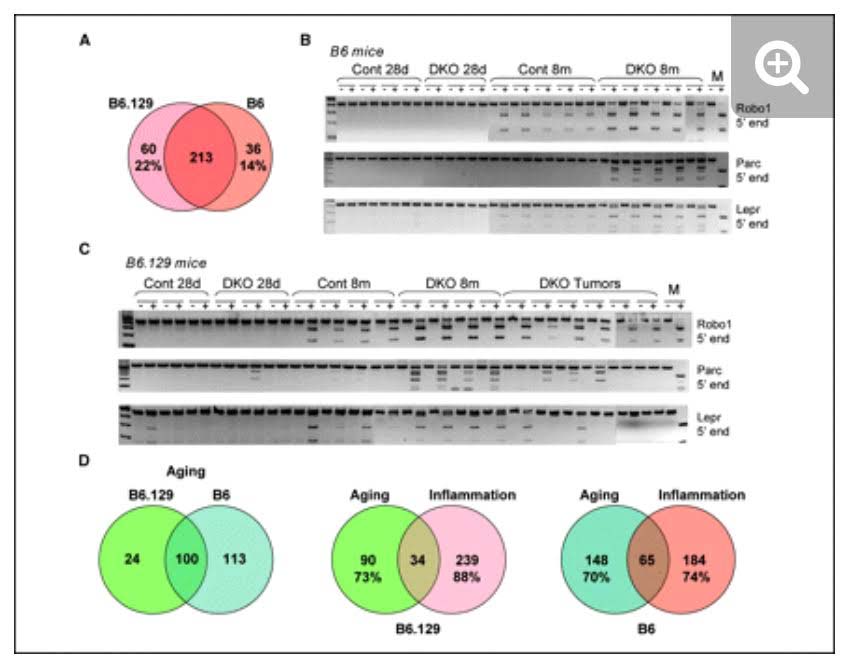
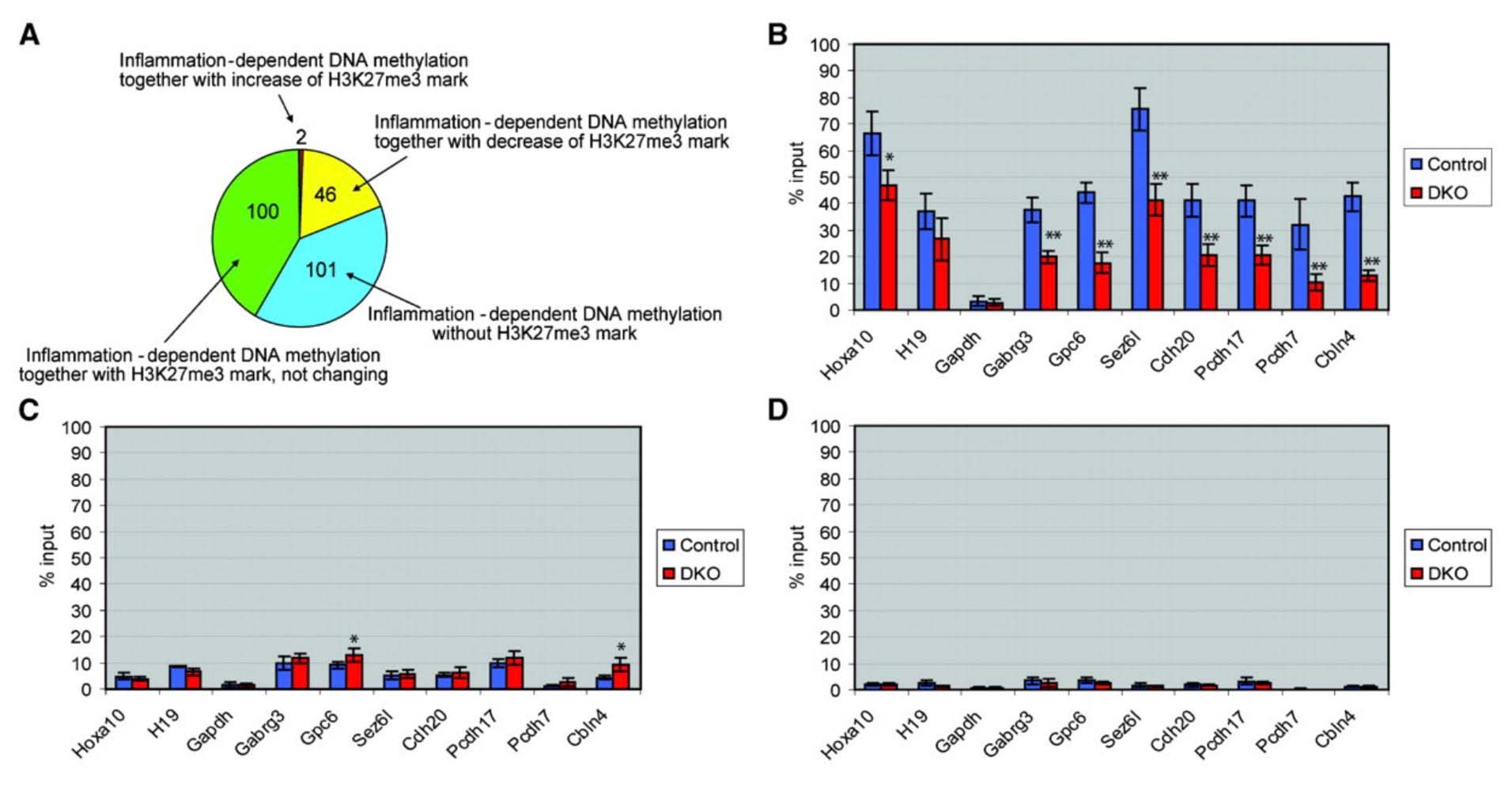
Reference and images source: 2008 Methylation of Polycomb Target Genes in Intestinal Cancer Is Mediated by Inflammation
11. Age-associated hypermethylation occurs preferentially at “Bivalent Chromatin Domains”
(To properly understand this subsection, please read it and then go back and read #10 above. The concept of bivalent chromatin domains and Polycomb protein target genes have a significant amount of overlap, and may actually be two ways of understanding the same subject).
We have discussed this point before. Rakyan and colleagues showed in 2010 that another specific feature of the age-associated hypermethylated sites was that they frequently occurred primarily in promoters with bivalent chromatin domains. Unlike Steve Horvath, they used cell-sorted CD14+ monocytes and cell-sorted CD4+ T cells. As did Steve Horvath, they used a computer program to pick out age-associated CpG sites that were either hypermethylated or hypomethylated with aging (aka differential methylation). They came up with a list of 360 CpG sites that underwent consistent, locus-specific CpG differential methylation. 147 of these underwent hypomethylation with aging and 213 underwent hypermethylation with aging. When they looked at the set of 213 age-associated hypermethylated CpG sites, they found 28 that were frequently hypermethylated in human cancer.
The specific bivalent chromatin marks were H3K27me3 and H3K4me3. The H3K27me3 is a mark of gene repression, whereas the H3K4me3 mark is a mark of gene expression. This is why they are called “bivalent chromatin” (both on and off chromatin marks). The H3K27me3 (repressive) mark is intertwined with the binding of a specific repressive Polycomb protein called “PRC2”. Thus both histone proteins and polycomb proteins help suppress gene expression at this bivalent chromatin domain regions. However the H3K4me3 mark is one for gene expression, thus these promoters are referred to as being “poised for action”. This type of chromatin is often seen in neural stem cells.
Reference:
2016 Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains
Here is an illustration of what a gene with bivalent chromatin domains is:
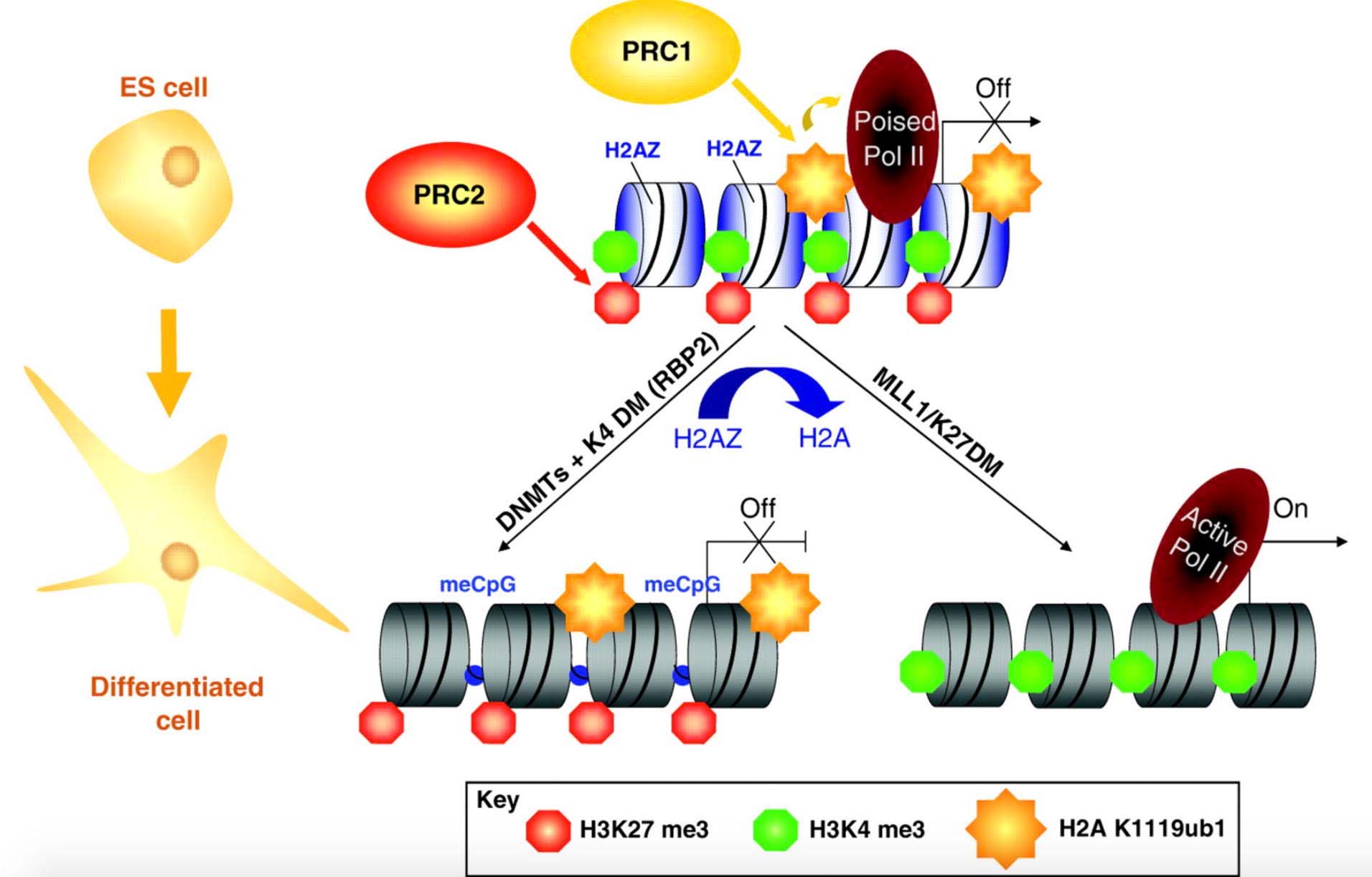
Reference and illustration source: 2009 Recruitment of Polycomb group complexes and their role in the dynamic regulation of cell fate choice
Here are two more illustrations of bivalent chromatin domains:
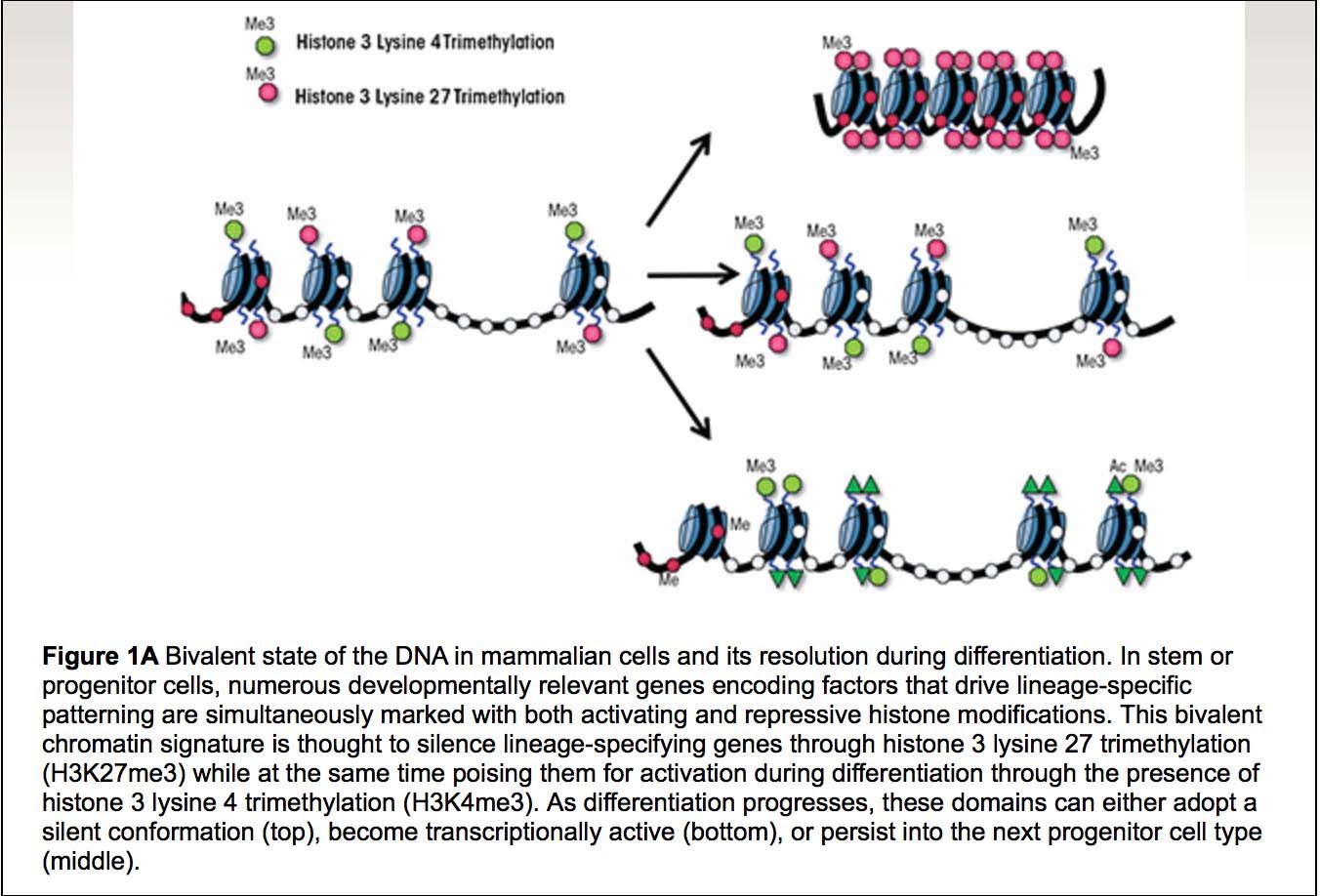
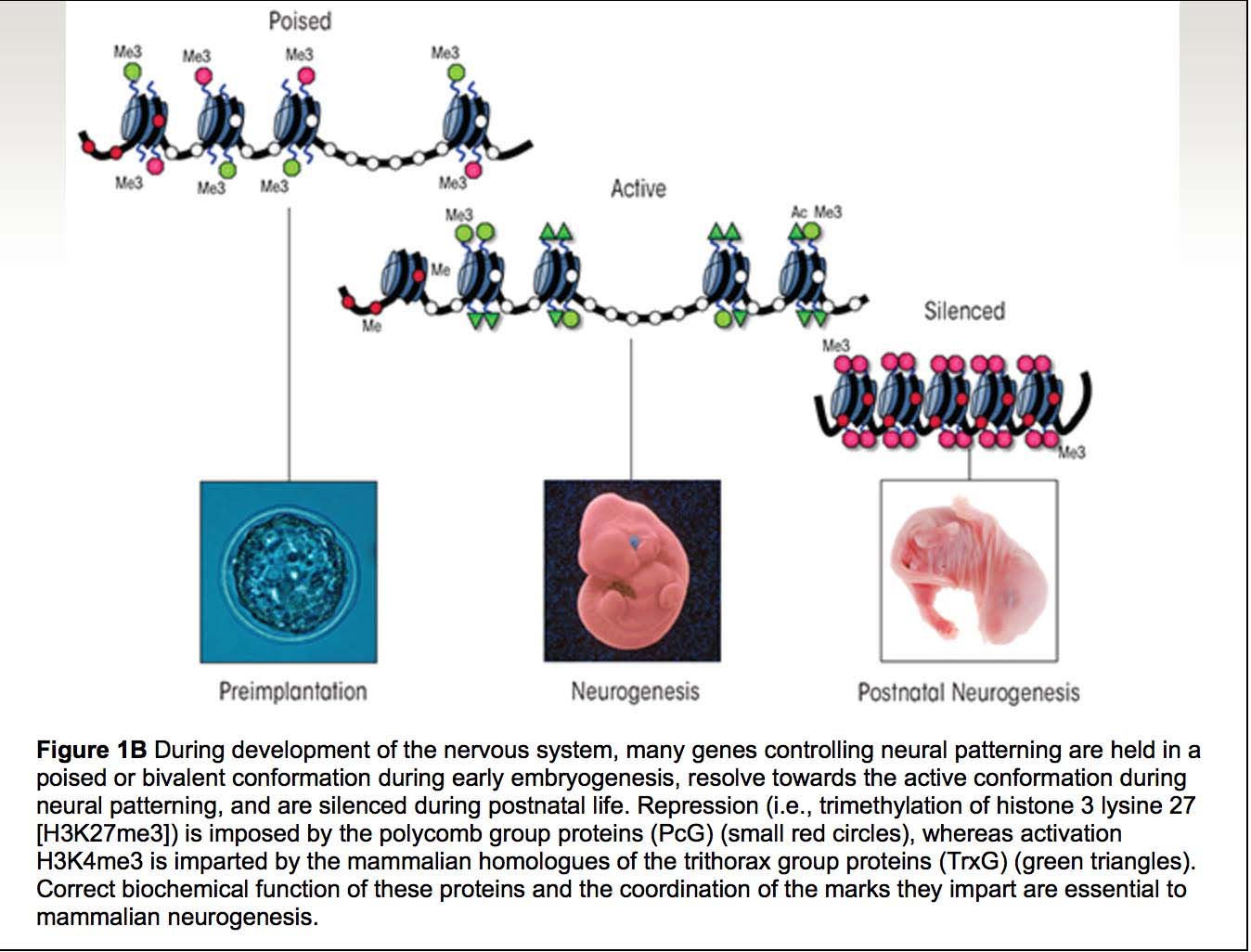
Images source: Prenatal Alcohol Exposure and Cellular Differentiation: A Role for Polycomb and Trithorax Group Proteins in FAS Phenotypes?
12. Chronic stress can lead to accelerated aging via age-assocated DNA methylation
This finding is based on a paper published by Anthony Zannas and colleagues from Max Plank Institute, as well as others from Duke University, Emory University, University of Miami, Queensland University, etc. in the 1st reference below, Anthony Zannas is explaining his research paper in easy-to-understand terms. The research group showed that 85 of Steve Horvath’s “DNA methylation clock” CpGs were found within glucocorticoid receptor (GR) response elements (GREs), which of course are the sites where cortisol/GR binds to the promoter or enhancer of a gene. They also found that chronic lifetime stress induced changes in gene DNA methylation and when exogenous dexamethasone (a powerful synthetic cortisol agonist) was given, it caused widespread changes in 110 of the 353 “DNA methylation clock” CpGs, as described by Steve Horvath. The dexamethasone also induced changes in gene expression of many genes near these “clock CpGs” (139 of 170 adjacent genes). Conclusion: 85 of the 353 CpG sites described in Steve Horvath’s “DNA methylation clock” are sites where cortisol, bound to the cortisol receptor (GR), activates gene expression. Hypermethylation of these “clock CpGs” occurs with aging and is accelerated by childhood stressors (child abuse) and chronic stress during adult life. Thus stress induces a state of “cortisol ineffectiveness”, thereby allowing chronic inflammation to occur, unabated by anti-inflammatory effect of circadian cortisol.
Thus too much stress changes the epigenome via hypermethylation of gene promoters and global hypomethyation of the genome, especially SINE repetitive elements (see last reference below). Also, it appears that this effect starts early in life and is lifelong cumulative From 2015 Do stress factors alter DNA methylation during aging? “The main glucocorticoid in humans is cortisol, which binds to and activates glucocorticoid receptors that act as transcription factors. Specifically, these receptors bind to specific DNA response elements and regulate the expression levels of a large number of target genes. Interestingly, glucocorticoid receptors not only affect gene transcription, but upon binding to target genes they can also change their DNA methylation state, and in some cases these changes can last long after cessation of the stressor. What were your main findings? We examined a highly traumatized cohort of African American individuals and found that exposure to more stress throughout the lifetime was associated with accelerated epigenetic aging. This effect was not seen with only childhood or recent stress and the effects were most pronounced in older individuals, so it appears that stress exposure accumulates to eventually affect the epigenome as one grows older. This effect was also more evident for personal stressors – stressors that affect the individual directly, for example; divorce, unemployment, and financial stressors. Whereas it was much weaker for network stressors – stressors affecting the individual’s social network, such as knowing someone who was robbed. Moreover, we also found that many of the age-related DNA methylation sites used to calculate epigenetic aging are located at glucocorticoid binding sites and undergo changes in methylation when individuals are exposed to a synthetic glucocorticoid, called dexamethasone. So it could be that high levels or dysregulated cortisol secretion in individuals exposed to more stress are driving these effects on epigenetic aging.”
References:
2015 High cortisol in 5-year-old children causes loss of DNA methylation in SINE retrotransposons: a possible role for ZNF263 in stress-related diseases “Here, we studied a homogenous group of 48 5-year-old children. By combining hair cortisol measurements (a well-documented biomarker for chronic stress), with whole-genome DNA-methylation sequencing, we show that high cortisol associates with a genome-wide decrease in DNA methylation and targets short interspersed nuclear elements (SINEs; a type of retrotransposon) and genes important for calcium transport: phenomena commonly affected in stress-related diseases and in biological aging. More importantly, we identify a zinc-finger transcription factor, ZNF263, whose binding sites where highly overrepresented in regions experiencing methylation loss. This type of zinc-finger protein has previously shown to be involved in the defense against retrotransposons.”
Also, methylation stress impact could be in some measure due to cortisol production associated with dysregulation of circadian sleep patterns. See Item 2 in Section C which follows.
SECTION C – CAUSES FOR THE TWO FACES OF DNA METHYLATION WITH AGING AND CANCER
- CpG Island promoters become hypermethylated with aging
About 60% of the protein coding genes in the human genome (19,000 genes) have a sequence called a “CpG islands”, or CGIs for short. These segments of cytosine-phosphate-guanine DNA bases are present in high numbers in DNA sequences that are between 500 and 2,000 base pairs (bp) long. Whereas in young, un-aged cells, these CpG islands are largely demethylated, with aging a subset of genes with CpG islands (CGIs) undergo hypermethylation. In cancer, the genes that undergo CpG hypermethylation in cancer include MLH1, MGMT, BRCA1, p16INK4a, p15INK4b, and Rb. With aging, different genes undergo CpG island hypermethylation. As a consequence some authors have subcategorized these 2 different types of CpG island methylator phenotypes into Type A and Type C methylator types. Type A CpG island methylation is seen with aging and is seen in noncancerous cells. Type C CpG island methylation is what is seen with cancer and is also referred to as CpG island methylator phenotype (CIMP). This CIMP pattern accounts for a specific subset of cancers where tumor suppressor genes are silenced by methylation, not by gene mutation, deletion, or chromosomal loss. In colorectal cancer, the CIMP subtype accounts for most of the sporadic cases of colorectal cancers that display micro satellite instability. This is mostly due to the CpG island hypermethylation of the MLH1 gene, which is a key gene in mismatch repair.
References:
Alcohol, DNA Methylation, and Cancer
1999 CpG island methylator phenotypes in aging and cancer
2015 Aging and DNA methylation
It appears that inflammation may be responsible for this age-related DNA hypermethylation face, that is hypermethylation of promoter sites of GpC islands . Specifically, the inflammatory cytokine IL-6 “drives” age-related epigenetic hypermethylation (at least of the p53 promoter),
I have always wondered what the driving force behind DNAm hypermethylation was. The paper cited below answers this question for at least one gene, a very important one – p53. The promoter for p53 has a CpG island and IL-6 inflammatory signaling silences the promoter of p53 via DNMT1 . Specifically, IL-6 signaling “drives” the gene expression of DNMT1, which then drives promoter hypermethylation of tumor suppressor CpG islands (at least for p53). This is a very important piece of the DNA methylation puzzle of aging. Since IL-6 is a cytokine biomarker for both aging and cellular senescence, this makes perfect sense and answers a lot of questions. It also answers or provides a direct link of how chronic inflammation and aging both result in cancer (by IL-6 mediated epigenetic silencing tumor suppressor gene expression). We know that JAK/STAT pathway signaling is triggered by IL-6 and that gene expression of many SASP genes are triggered by IL-6 mediated JAK/STAT3 signaling. This pathway drives both cancer and aging! This was a true “Ah-Ha moment” for me, along with #8 below. Here is the article on this.
Reference: 2005 Interleukin 6 supports the maintenance of p53 tumor suppressor gene promoter methylation
- Circadian cortisol secretion (HPA axis) appears to play a major role in the DNA hypomethylation face of “epigenetic aging”.
Cortisol induces hypomethylation by binding to glucocorticoid receptors (GR), suppressing DNMT3a, and activating TET3. As we discussed before, glucocorticoid receptor activation of the TET3 demethylase enzyme “Drives” DNA methylcytosine oxidation in a 3 step reaction from 1) methylcytosine to hydroxymethylcytosine, abd then to 2) carboxy methylcytosine, and then to 3)formylcytosine, and ultimately to unmethylated cytosines. This cortisol effect simultaneously induces global genome CpG hypomethylation and prevents DNMT3a-mediated methylation of DNA other than CGIs. Thus cortisol/GR binding to the glucocorticoid response element (GRE) induces both focal DNA hypomethylation at CGIs and global hypo-methylation throughout the entire genome by activating TET3, which demethylates methylcytosines.
References:
2014 TET enzymes and DNA hydroxymethylation in neural development and function — How critical are they?
Methyl cytosines can also undergo spontaneous deamination, resulting in the cytosine being converted into a thymine. This spontaneous deamination is not as common as TET-mediated loss of cytosine methyl groups, however. The consequence of cortisol-induced TET3-mediated CGI demethylation includes the re-expression of normally silenced repetitive elements (Ex: LINE-1), the expression of normally silenced pseudogenes, and the expression of long-noncoding RNA and microRNA genes with glucocorticoid response elements (GRE) in their promoter or enhancer regions.
Sleep patterns and stress seem to be involved here. Above in Item 12 in Section B we discussed the role of stress and cortisol concentration in having a cumulative lifelong impact on DNA methylation. Cortisol concentration is normally strongly effected by a circadian pattern as is illustrated in this graph:
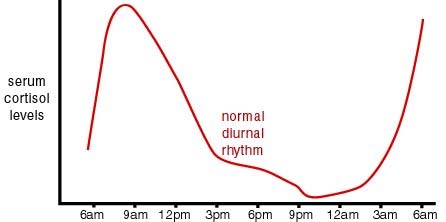
Image and legend source: “Cortisol has a very distinct circadian rhythm that is regulated by the central clock located in the hypothalamus. In a normal circadian cycle, also known as the cortisol curve, you have very low cortisol levels around midnight; from there, levels start to build up overnight to peak first thing in the morning. From there, cortisol levels slowly decline throughout the day [9]. This natural curve can become easily disrupted, however, when distress or stressors are added to your daily routine [11]. When you experience stress of any kind, your adrenals are signaled by the pituitary gland to secrete cortisol. Unfortunately, we are not superheroes and there is a limit to how much stress our bodies can handle. When many different life stressors become too much for your body to handle, our adrenals work overtime to try to mobilize your our body’s response to stress (whether it’s physical, emotional or psychological) by secreting more and more cortisol into your system.”
Thus, stress dysregulation of circadian rhythms may profoundly influence cortisol production inducing DNA hypomethylation that is cumulative with aging. Stresses can include worries that keep people up at night, night shift work and frequent changes of time zones.
Next we take a slight detour and discuss the role of TET enzymes in global demethylation
3. Methylated cytosines can undergo demethylation by proteins called “TET enzymes” triggered by cortisol
Although the enzymes that methylate DNA were discovered first and initially, were thought to be a “one-way reaction”, now it is clear that cytosines can undergo both methylation and demethylation. Demethylation is a key feature of both normal cell biology as well as pathological conditions such as cancer. DNA cytosine demethyation can be active or passive. Passive demethylation occurs when DNA is replicated during the S-phase of the cell cycle. Normally, an enzyme called DNMT1 methylates the new strand of DNA that is formed, using the old strand as a “methylation template”. However, if there is a deficiency of methyl donors due to folate deficiency, B12 deficiency, or a inadequate protein intake during pregnancy, the new strand of DNA may not be methylated appropriately by DNMT1. This is called “passive demethylation” and is due to vitamin deficiencies or maternal protein-calorie malnutrition during pregnancy.
Active demethylation occurs by enzymatic oxidation of the methyl group on methyl cytosine. This is catalyzed by a family of enzymes called “ten-eleven translocation” enzymes, or TET for short. There are three TET enzymes in humans, TET1, TET2, and TET3. All three of these can demethylate DNA. TET3 is the enzyme that is triggered by cortisol binding to the glucocorticoid receptor.
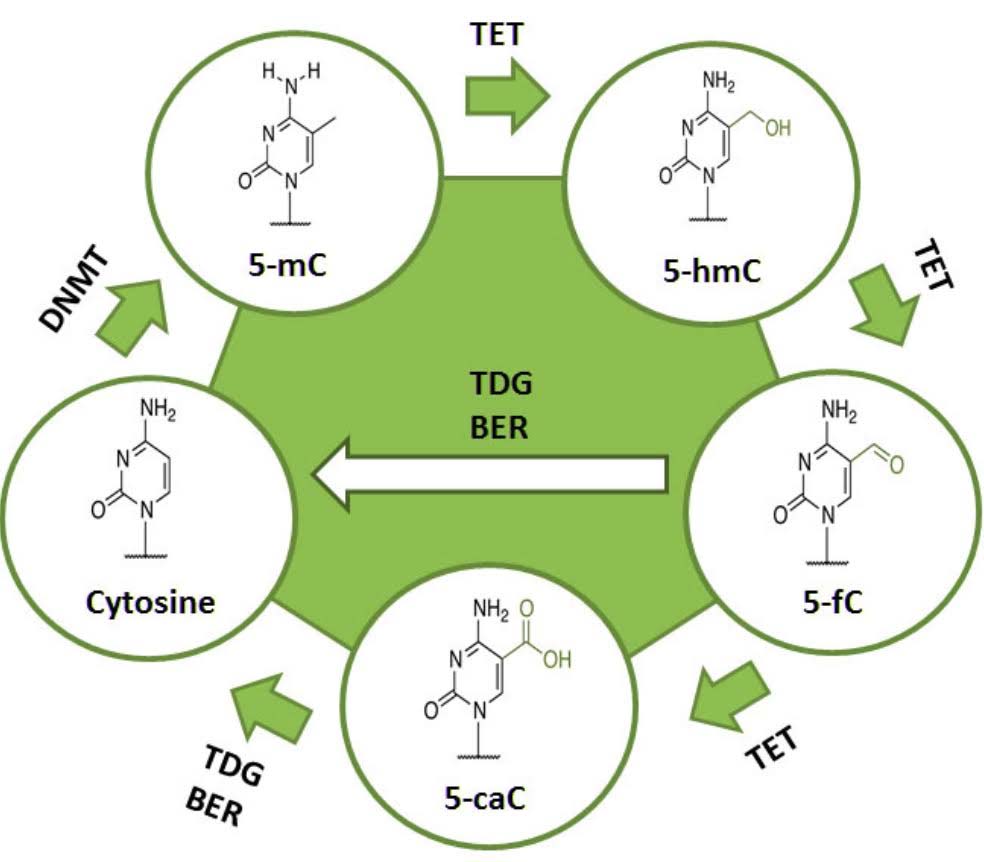
4. Global DNA demethylation is a physiologically normal reaction that must occur twice during early embryogenesis. It also must occur with normal erythropoiesis, but not globally (only 1/3rd of the genome is demethylated). Thus Global DNA demethylation should not be viewed merely as something “bad,” associated only with aging.
Although the demethylation of DNA by TET enzymes has mostly been thought of as being something “bad”, it is actually an evolutionarily-conserved feature of normal embryogenesis, erythropoiesis, and other phenomena in stem cells. Twice during early embryologic development, in the zygote and in primordial germ cells, a global loss of CpG methylation occurs in the entire genome. Methylation is re-established at the blastocyst stage of embryogenesis and remains globally stable throughout somatic cell development.
A similar global demethylation of DNA occurs at the onset of erythroid transcription in early erythroid precursors. This event triggers a global demethylation of DNA, much like what happens in early embryogenesis. However with erythropoiesis, only 1/3rd of the genome is demethylated, including enhancers, promoters, and repetitive elements. This occurs in the bone marrow. Here is an illustration of this phenomena.
Reference: 2012 Global DNA Demethylation During Erythropoiesis in vivo
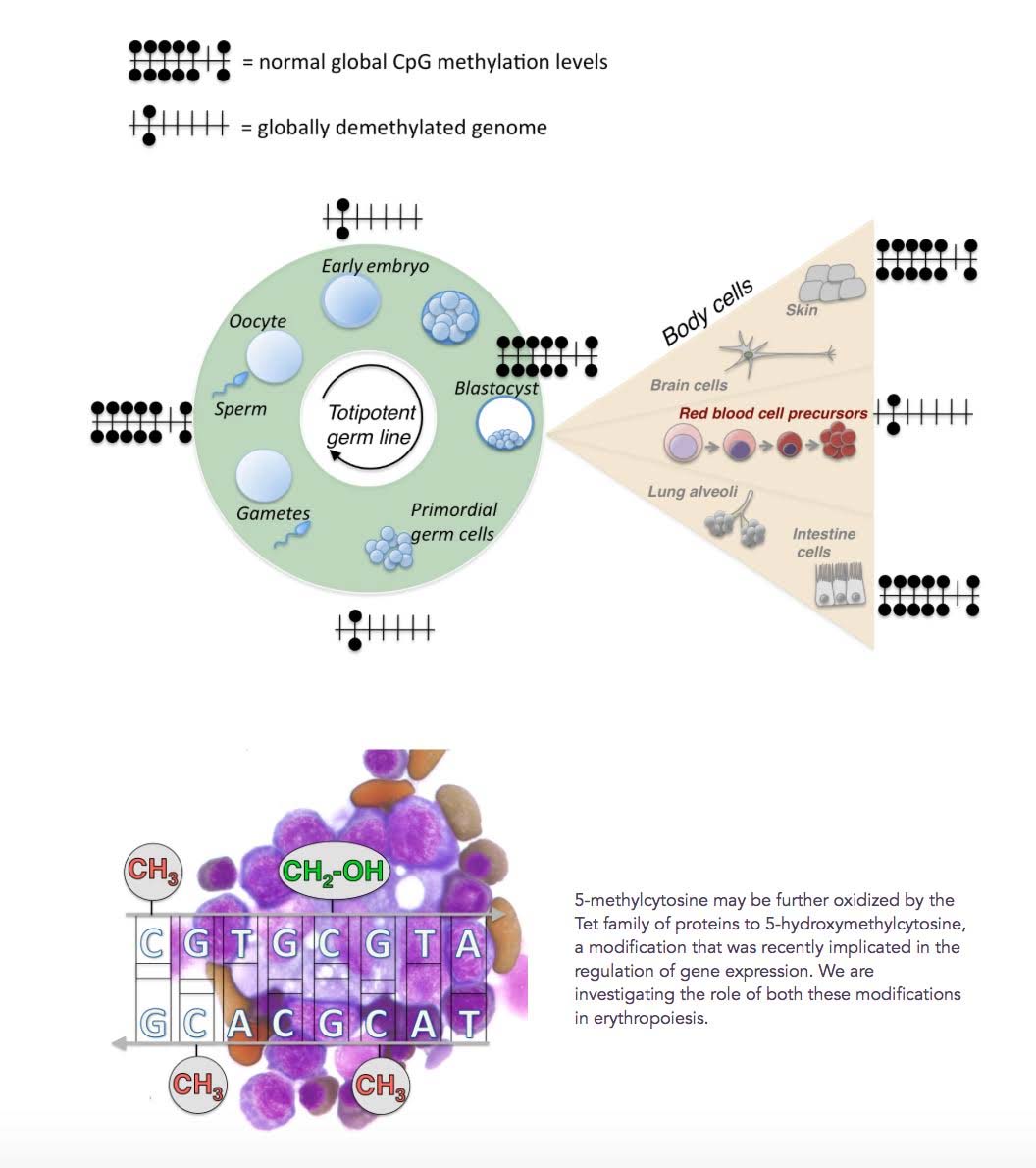
Illustration source: Genome-wide DNA demethylation in Erythropoiesis
“ Approximately a third of all DNA methylation is lost from essentially all genomic elements, including enhancers, promoters and repetitive elements. Global demethylation is intrinsic to erythroid differentiation, taking place in yolk sac, fetal and adult erythropoiesis including the human bone-marrow.”
- LINE-1 repetitive DNA repression is lost with aging (but not due to CpG hypomethylation)
LINE-1 methylation is lost with chronic alcohol abuse, however.
The only repetitive DNA type found in the human genome that can still undergo active transposition is the LINE-1 (L1) retrotransposon. 1/6th of the entire human genome is made up of LINE-1 retrotransposon DNA (over 500,000 copies), but most all of these are nonfunctional copies. The only subclass of L1 retrotransposons that can still function as a “self copy machine” is the human-specific L1HS-Ta subfamily of LINE-1. In the L1HS-Ta subfamily only 100-150 copies work.
However, all of the LINE-1 copies are normally suppressed by DNA hypermethylation and heterochromatin. Whereas many experts assumed that LINE-1 DNA becomes hypomethylated with aging, no evidence for this exists to date. However there is clear evidence that LINE-1 retrotransposons are expressed at increasing frequency with aging. Thus there must be some other mechanism besides CpG hypomethylation to explain the re-expression of L1 transposable elements. Recent discoveries have linked the re-expression of LINE-1 to the loss of SIRT6 activity. As it turns out, SIRT6 is an enzyme that must “multi-task”. It must silence LINE-1 (its normal “day job”) and it also is recruited to DNA damage to help in double stranded DNA breaks.
Unfortunately it cannot do both of these “multi-tasks” simultaneously. Also with aging there is a decrease in NAD+ within the cell nucleus. NAD+ is a required, consumed cofactor for SIRT6 activity. As a consequence of these factors, there is a well-described, consistently-seen increase in LINE-1 transposable element expression with aging. See our blog entry Transposable DNA elements –- Part 2: The Self-copy Machines in Your Genes.
Here is a graph showing the increase in expression of transposable elements (L1, B1, and B2) in the liver and muscle of rodents:
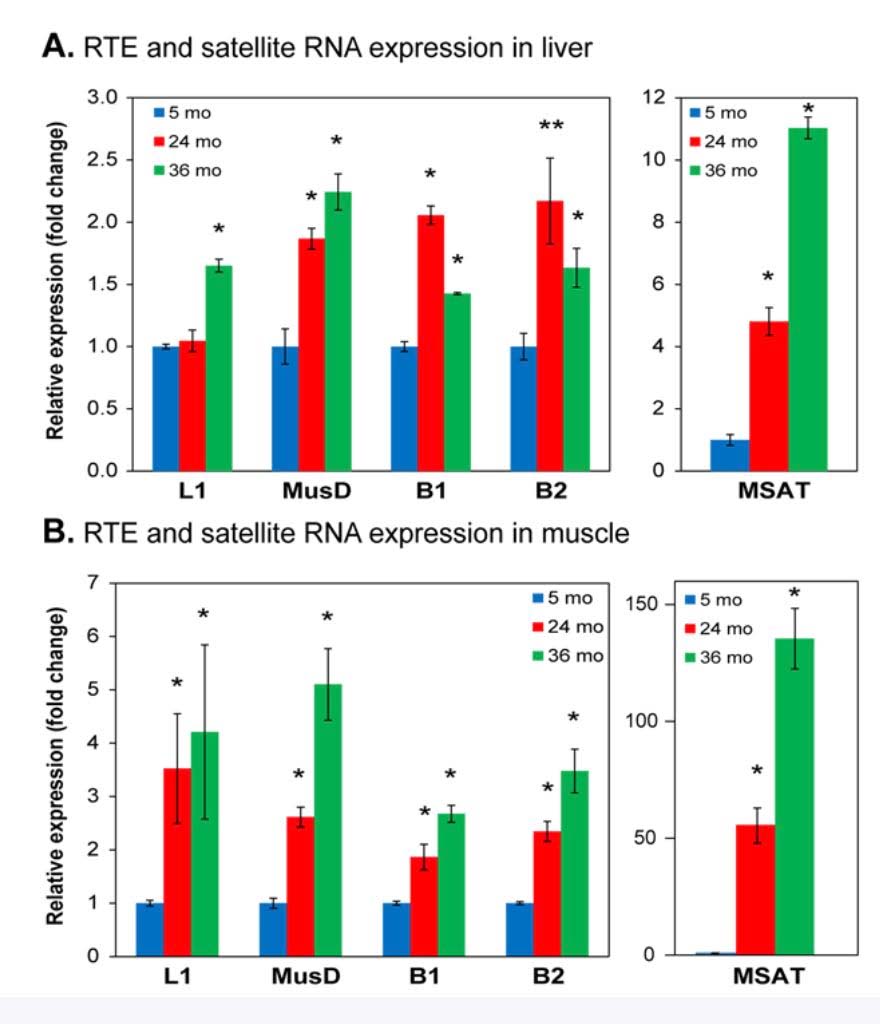
As you can see, there is a dramatic increase in expression, especially in the liver. SIRT6 plays a key role in suppressing L1 expression by a completely different mechanism than DNA CpG methylation or Histone based silencing of L1. Instead, SIRT6 binds to the 5’-UTR region of the L1 promoter, where it monoribosylates KAP1, a nuclear co-repressor protein. This allows KAP1 to interact with HP1a, the protein that forms heterochromatin. Unfortunately, during the course of aging, SIRT6 has to “fly away” and help repair DNA damage. While SIRT6 is not “at home”, repressing L1, the L1 gene can be transcribed leading to a “copy machine” that can copy the L1 retrotransposon, or can be “hijacked” and used by other repetitive elements like Alu repeats. These active “copy machines” can also copy protein-coding genes, such as APP (amyloid precursor protein), which plays a major role in Alzheimer’s disease.
References:
2010 Distinctive patterns of age-dependent hypomethylation in interspersed repetitive sequence
2013 Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues
2016 Activation of individual L1 retrotransposon instances is restricted to cell-type dependent permissive loci
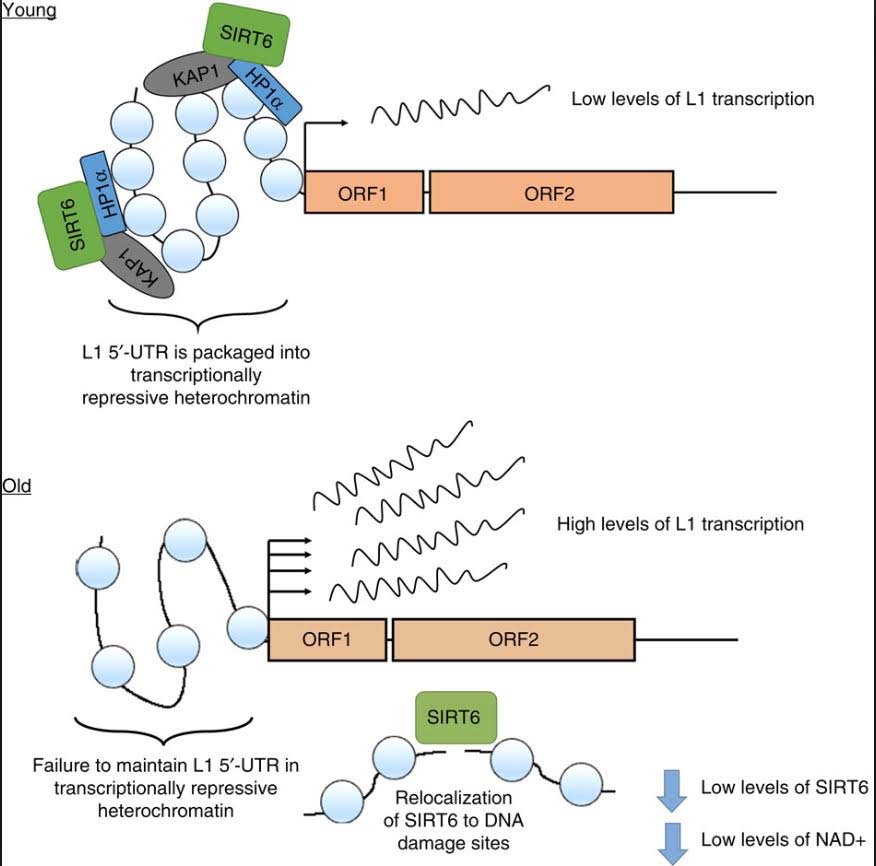
Reference and image source: 2014 SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age
Conclusion: LINE-1 retroelements are normal silenced by DNA CpG methylation, histone-based silencing, heterochromatin formation, and one unique mechanism mediated by SIRT6 monoribosylation of KAP1. With aging, NAD+ levels decline and SIRT6 gene expression declines. In addition to its role in LINE-1 silencing, SIRT6 must also help with DNA repair. As a consequence, there is a failure to maintain a L1- 5’-UTR repressive chromatin state. This results in a dramatic increase in expression of L1 retroelements with aging. For background on this see the blog entries NAD+ an emerging framework for life health and life extension — Part 2: Deeper into the NAD World, hopeful interventions and Transposable DNA elements –- Part 2: The Self-copy Machines in Your Genes.
More references:
- Weisenberger, D.J. et al. (2005) Nucleic Acids Res., 33(21): 6823-6836.
- Lisanti, S. et al.(2013) PLoS One, 8(11): e79044.
- Kazazian, H.H.J. (2000) Science, 289(5482): 1152-1153.
- Yang, A.S. et al.(2004) Nucleic Acids Res., 32(3): e38.
- Ogino, S. et al.(2008) Journal of the National Cancer Institute, 100(23): 1734-1738.
- Cho, Y.H. et al.(2010) (2010) Anticancer Research, 30(7): 2489-2496.
- Irahara, N. et al.(2010) Journal of Molecular Diagnosis, 12(2): 177-183.
- Wilhelm, C.S. et al.(2010) Clinical Cancer Research, 16(5): 1682-1689.
- Baccarelli, A. et al.(2010) Epidemiology, 21(6): 819-828.
- Kim, M. et al.(2010) PLoS One, 5(3): e9692.
- Ohka, F. et al.(2011) PLoS One, 6(8): e23332.
6. Alu repeat and HERV-K expression occurs with aging due to the loss of CpG methylation.
Whereas loss of SIRT6 activity is the main reason why LINE-1 transposable elements become activated with aging, the loss of DNA CpG methylation is the main reason why SINE-1 repetitive elements increase in expression with aging. The same is true for Human Endogenous Retroviruses (HERV). Since the main transcriptionally active subtype of HERV transposable elements is the HERV-K subtype, I will refer to this specific subcategory of HERVs in this review. Here is some data that shown how LINE-1 demethylation does not correlate with aging, whereas SINE-1 demethylation (Alu repeats) and HERV demethylation do increase with aging.
Reference: 2010 Distinctive patterns of age-dependent hypomethylation in interspersed repetitive sequences
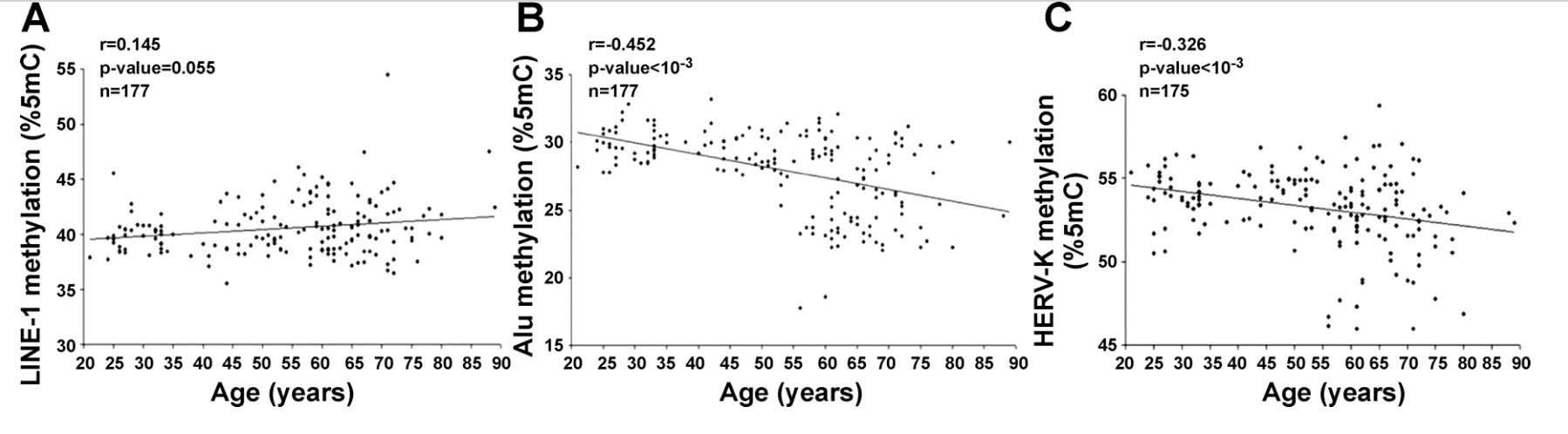
Image source: 2010 Distinctive patterns of age-dependent hypomethylation in interspersed repetitive sequences
7. Satellite DNA (pericentric repeats, etc.) become hypomethylated with aging and are actively transcribed
Satellite and micro satellite repeats are another form of repetitive elements that play a key role in aging and disease, but are not transposable elements.
8. Telomeric DNA silencing is lost (not due to DNA methylation) and the telomeric repeats are transcribed into TERRA
Another feature of aging is the transcription of several subtelomeric loci onto the telomeric DNA itself. The RNA molecules made by this phenomena is a long non-coding RNA that has been named “TERRA”, which stands for “Telomeric repeat-containing RNA (TERRA). TERRA binds to the Sheltering protein, TRF2, and facilitates the formation of heterochromatin. See our blog entry Nuclear Aging: The View from the Telomere end of the Chromosome – Part 3 – Telomere Molecular Biology and GUT implications – The two faces of P53.
9. ROS-induced DNA damage and Base excision repair mechanisms also contribute to age-related DNA damage.
This is the last “Ah-Ha moment” for me. Totally expected, of course, but I still had not figured this out this completely as it fits together with the other elements described here.
Reference: 2014 The Emerging Nexus of Active DNA Demethylation and Mitochondrial Oxidative Metabolism in Post-Mitotic Neurons “The variable patterns of DNA methylation in mammals have been linked to a number of physiological processes, including normal embryonic development and disease pathogenesis. Active removal of DNA methylation, which potentially regulates neuronal gene expression both globally and gene specifically, has been recently implicated in neuronal plasticity, learning and memory processes. Model pathways of active DNA demethylation involve ten-eleven translocation (TET) methylcytosine dioxygenases that are dependent on oxidative metabolites. In addition, reactive oxygen species (ROS) and oxidizing agents generate oxidative modifications of DNA bases that can be removed by base excision repair proteins. These potentially link the two processes of active DNA demethylation and mitochondrial oxidative metabolism in post-mitotic neurons. We review the current biochemical understanding of the DNA demethylation process and discuss its potential interaction with oxidative metabolism. We then summarise the emerging roles of both processes and their interaction in neural plasticity and memory formation and the pathophysiology of neurodegeneration. Finally, possible therapeutic approaches for neurodegenerative diseases are proposed, including reprogramming therapy by global DNA demethylation and mitohormesis therapy for locus-specific DNA demethylation in post-mitotic neurons.”
Yes the Denham Harman 1950’s oxidative damage theory of aging seems to have died years ago, but it also keeps arising from the dead in new contexts such as this one of DNA demethylation.
SECTION D – FURTHER DETAIL ON CPGs AND CGIs
1. More on what is a CpG Island. CpG island = CGI – CpG islands are found in the promoter regions of 70% of mammalian genes
To familiarize our readers with the lingo of DNA methylation, we expand the definition given previously of a CpG island (abbreviated as “CGI”). A CGI is a sequence of “Cytosine-phosphate-Guanine” DNA dinucleotide repeats that are > 200 bp long and where at least 55% of DNA bases in the sequence are CpGs. Furthermore, to be a CpG island (CGI), the sequence must have an observed-to-expected ratio of > 65% CpGs. (The old definition was 50% CpGs and 60% observed-to-expected ratio, but this was changed in 2002). Not all genes have CGIs, but in mammals, 70% of protein coding genes have CGIs in their promoters. 70% of mammalian genes contain a CGI in their gene promoters. However, only 35% of all CGIs are found in the promoter region of genes. The other 65% of CGIs are found within repetitive elements (10%), introns (7.7%), the “head” of a gene (5′-UTR) and the “tail” of a gene (3′-UTR). CGIs in promoters are mostly unmethylated, whereas CGIs in introns are differentially methylated, changing their methylation state in a dynamic fashion, in response to various external stimuli, such as cortisol-glucocorticoid receptor binding.
The genes that have CGIs in their promoter regions include “house keeping” genes (which are constitutively expressed as well as some regulated genes that are only expressed at certain times. In addition to having a CGI in the promoter, there are two other features of mammalian promoters: 1) low nucleosome occupancy (no histone “spool” winding at these promoters), and the 2) pre-association of RNA Pol II at the promoter site in inactive genes.
As described here, the epigenetic silencing of genes by CGI hypermethylation play a major role in aging-associated diseases such as Type II Diabetes and Cancer. The other Half of the DNA methylation story is just emerging – the story of why gene promoter CGIs become hypermethylated with aging. In a nutshell Here is what we know and what we do not know: promoter hypermethylation of CGIs is not due to cortisol. Instead, it is likely to be due to things like foreign chemicals, alcohol, viruses, and other invaders.
2. CpG islands are mostly unmethylated, whereas CpG dinucleotides found outside of GpG islands are mostly methylated – on “R loops”
This is a fundamental feature of CGIs – they are mostly unmethylated. Several molecular mechanism to explain why GCIs are resistant to methylation, including 1) “GC skew”; 2) “R loop formation”; and 3) the presence of RNA Polymerase II occupying the promoter region. (All of these three molecular mechanisms are linked and could be viewed as a single mechanism, as well). Not all promoter CGIs are “methylation resistant”. Methylation resistant CGIs within promoters display significant “strand asymmetry” in the distribution of cytosines and guanines immediately downstream from the transcription start site (TSS). This DNA feature is called “GC skew”. “GC skew” is a DNA feature found in 46% of all gene promoters with CGIs. If the mRNA produced from a region of GC skew is “G-rich” (i.e. G-rich mRNA), it forms an mRNA with a big loop called an “R loop”. Thus transcription of genes through a DNA region with “GC skew” results in a mRNA that undergoes “R loop formation”. R loop formation of the mRNA protects the CGI from DNMT3B1, the methyltransferase that normally methylates CpGs with no “GC skew” and no “R loop”.
Another molecular mechanism for “methylation resistant” promoters is due to the occupancy of RNA polymerase II (the enzyme that copies DNA). In many genes with CGIs in their promoter, RNA Pol II sits in the promoter region, poised to start transcription as soon as the signal for gene copying occurs. In this scenario, RNA Poly II occupies the promoter region, thereby physically blocking DNMTs. Below is an illustration of the formation of R loops and the subsequent blockade of DNMT3b.
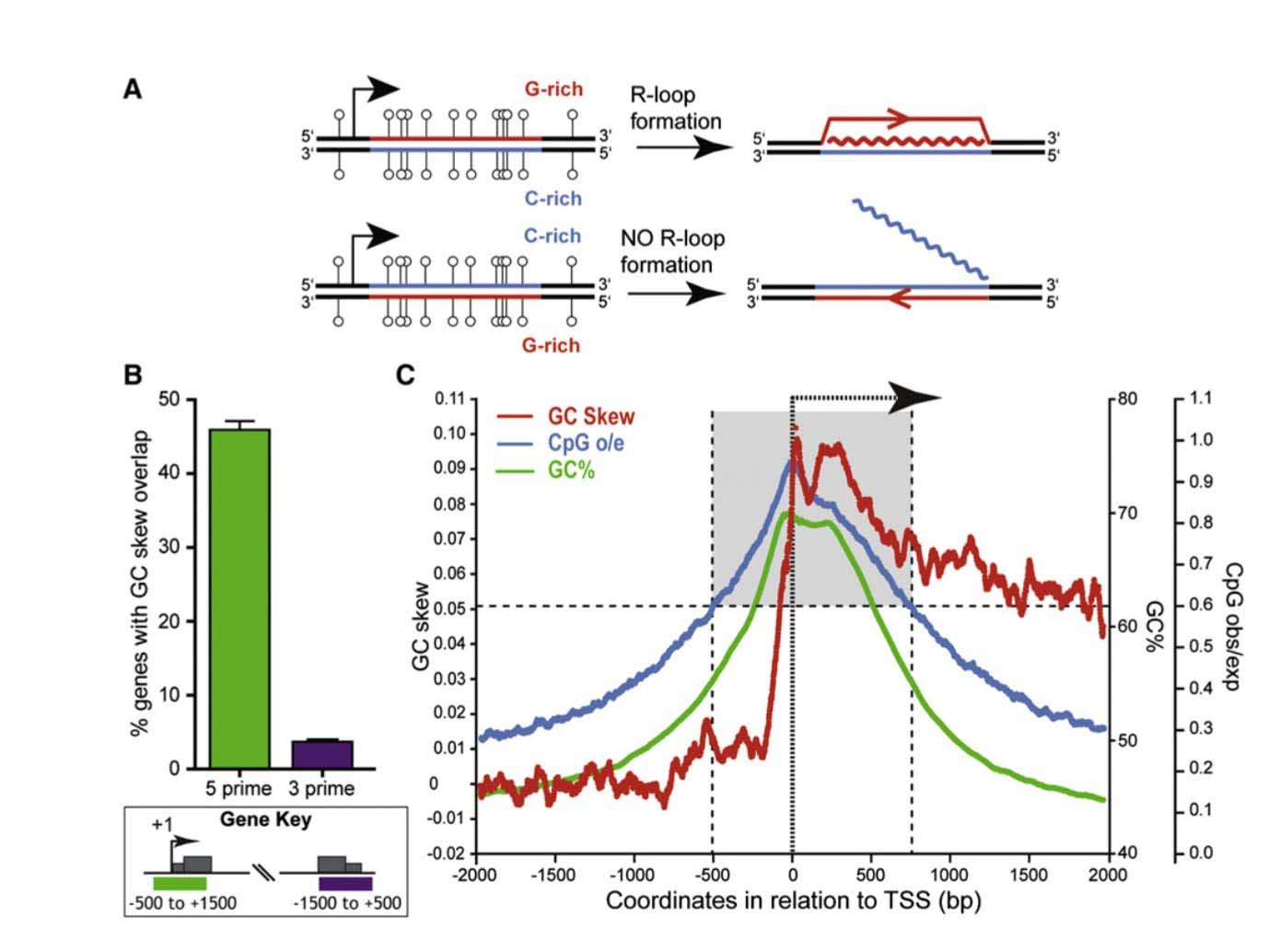
Reference and image source: 2012 R-Loop Formation Is a Distinctive Characteristic of Unmethylated Human CpG Island Promoters
- ” CpG island (CGI) promoters are characterized by GC skew downstream of TSS
- Transcription through skewed CGI promoters leads to R loop formation
- R loop formation potential is predictive of the unmethylated state of CGIs
- R loops may establish a protective chromatin environment against DNA methylation”
“Summary: CpG islands (CGIs) function as promoters for approximately 60% of human genes. Most of these elements remain protected from CpG methylation, a prevalent epigenetic modification associated with transcriptional silencing. Here, we report that methylation-resistant CGI promoters are characterized by significant strand asymmetry in the distribution of guanines and cytosines (GC skew) immediately downstream from their transcription start sites. Using innovative genomics methodologies, we show that transcription through regions of GC skew leads to the formation of long R loop structures. Furthermore, we show that GC skew and R loop formation potential is correlated with and predictive of the unmethylated state of CGIs. Finally, we provide evidence that R loop formation protects from DNMT3B1, the primary de novo DNA methyltransferase in early development. Altogether, these results suggest that protection from DNA methylation is a built-in characteristic of the DNA sequence of CGI promoters that is revealed by the cotranscriptional formation of R loop structures.”
3. CGI methylation is the primary way that specific gene copies are silenced by molecular mechanisms and this specific silencing is hereditable. When only the maternal or paternal copy of a gene is silenced, this phenomenon is called “imprinting”
From What is imprinting? “For most genes, we inherit two working copies — one from mom and one from dad. But with imprinted genes, we inherit only one working copy. Depending on the gene, either the copy from mom or the copy from dad is epigenetically silenced. Silencing usually happens through the addition of methyl groups during egg or sperm formation. The epigenetic tags on imprinted genes usually stay put for the life of the organism. But they are reset during egg and sperm formation. Regardless of whether they came from mom or dad, certain genes are always silenced in the egg, and others are always silenced in the sperm.”
The classic example of imprinting that is used in genetic textbooks is inactivation of the paternal copy of the X chromosome in females. Imprinting is clearly due to CGI DNA methylation of hundreds of genes on the inactive X chromosome. Specifically, only one copy of the gene is silenced with imprinting. Imprinting of genes also occurs on other chromosomes, albeit infrequently. Paternal imprinting means that the paternal copy of the gene is silenced by CGI methylation. Maternal imprinting means that the maternal copy of the gene is silenced by CGI methylation. Here is an illustration of DNA cytosine methylation.
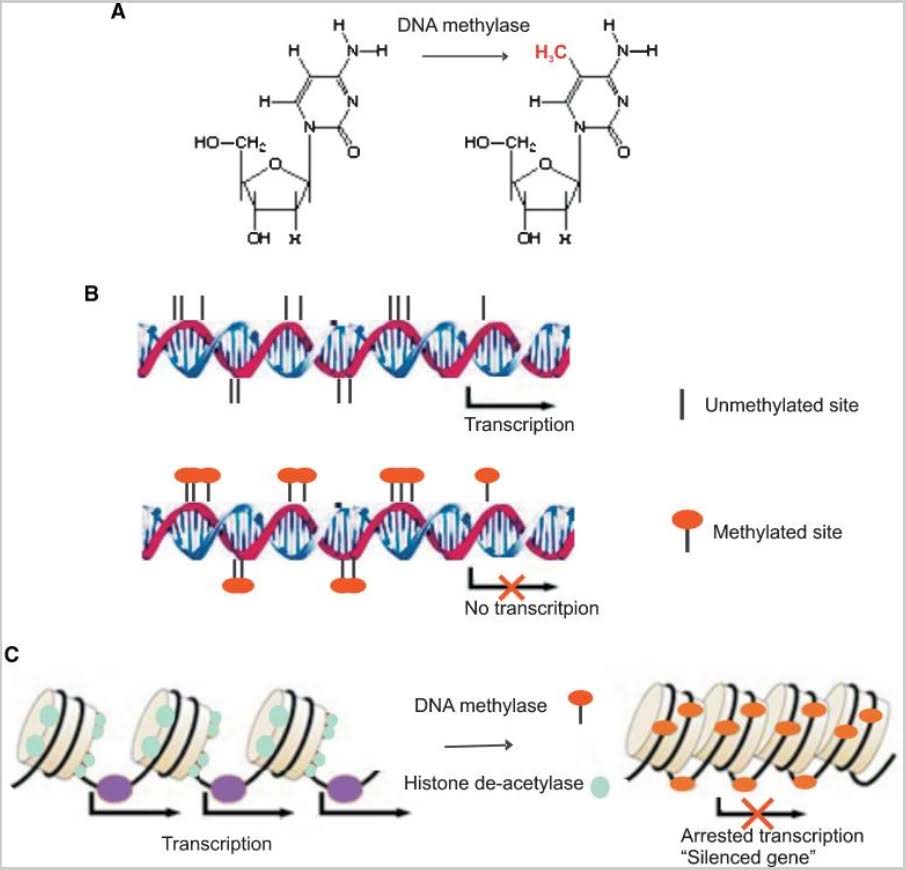
Illustration Ref: 2009 Epigenetic and microRNA-mediated regulation in diabetes
Schematic figure of epigenetic regulation mechanism. DNA methylase catalyze the transfer of a methyl group to the cytosine residues in CpG dinucleotide sequences (A). Methylation of the CpG islands in the gene promoter region inhibits gene expression (B). Histone acetylation plays an important role in the regulation of gene expression. Hyperacetylated chromatin is transcriptionally active whereas hypoacetylated chromatin is silent. Methyl-CpG-binding proteins interact with histone deacetylase causing gene silencing (C).
4. There are 37,729 CGIs in the human genome that are > 1,000 bp, many more than in mice
That is how many CpG islands there is in the human genome that are at least 1,000 base pairs in length (1 kb). Of course that does not mean all 1,000 nucleotides in the CGI are cytosine-phosphate-guanine (CpG) dinucleotides, but it basically means that there are MORE total CGIs than there are protein coding genes (the latest count is that there are between 19,000 and 20,000 protein coding genes in the human genome). Moreover, human genomes have a far greater number of CGIs than mice do, suggesting that the proliferation of this type of epigenetic gene regulation evolved (recently) since primates diverged from rodents in the “phylogenetic tree” of molecular evolution.
References:
2009 CpG islands or CpG clusters: how to identify functional GC-rich regions in a genome?
2010 Epigenetic regulation of the expression of genes involved in steroid hormone biosynthesis and action
5. 7. 35% of CGIs are in promoters
That is the percentage of the total number CpG islands in the human genome. But only 70% of all CGIs are associated with genes. (The rest are in intergenic regions, in “junk DNA” like pseudogenes and transposable elements. If you only look at the 70% that are associated with genes, over half of these are in the promoter region of the gene. In these promoter-associated genes, 56% of the CGIs overlap the transcription start site TSS). Thus it is clear that the primary purpose of promoter CGIs is to block the initiation of transcription by RNA pol II by methyl binding proteins (MBPs) that attach themselves to the methyl group and attract depressor complexes.
References:
2010 Epigenetic regulation of the expression of genes involved in steroid hormone biosynthesis and action
2009 CpG islands or CpG clusters: how to identify functional GC-rich regions in a genome?
6. A few of the CGIs in promoters are imprinted
To date, approximately 100 genes have been found to be imprinted. Of these, only a few undergo “allele-specific” DNA methylation of their promoter regions. The IGF2 gene and the H19 gene two examples of this phenomena. Here is an illustration of “allele-specific” DNA methylation of the IGF2 gene promoter sequence and the neighboring H19 gene.
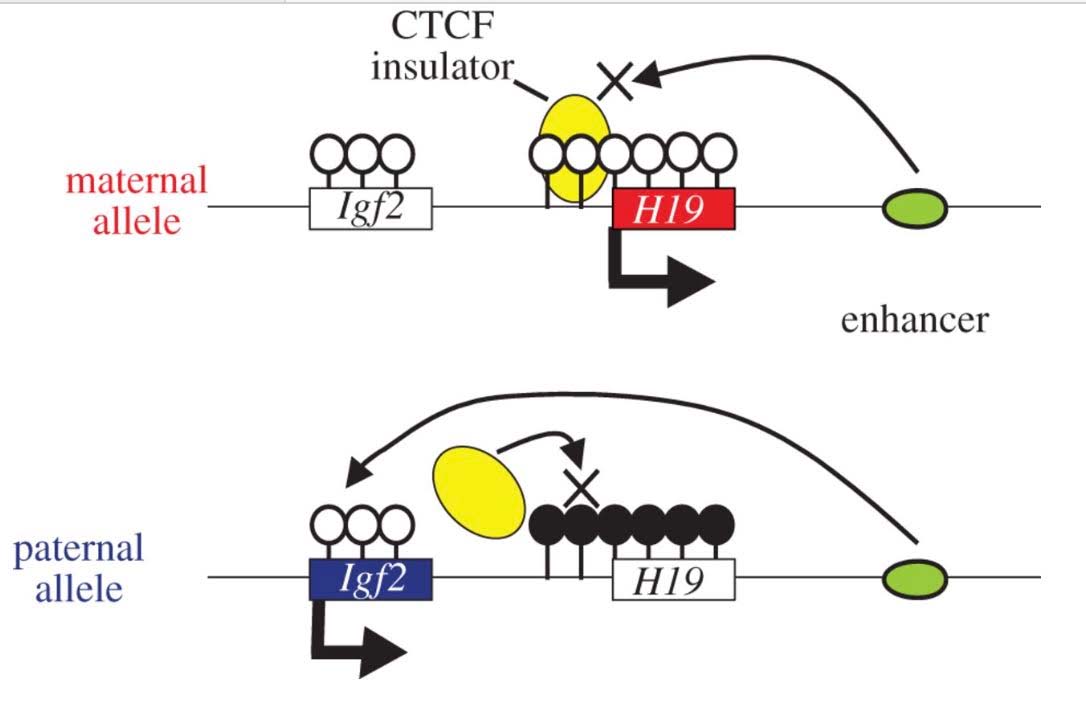
Reference and image source: 2012 The origin and evolution of genomic imprinting and viviparity in mammals
7. Other than CGIs found in the promoter region, CGIs are also found in the 5’ UTR region of genes.
The region of a gene that is “upstream” from the promoter is called the “5′ untranslated region” (5′-UTR). This region of a gene does not have any transcription factor binding sites (I.e. also called “response elements”) but the 5′-UTRs still has important regulatory roles in gene expression, including binding sites for depressor plexes. Thus it is not a surprise that CpG islands are found in these 5′-UTR regions.
References:
2009 CpG islands or CpG clusters: how to identify functional GC-rich regions in a genome?
2006 CpGcluster: a distance-based algorithm for CpG-island detection
2009 CpG islands: algorithms and applications in methylation studies
8. Some CGIs are found in the 3′-UTR region (“tail”) of the gene
In 1993, Bird and colleagues described the finding of CGIs in the “tail” of a gene, referred to as the 3′-untranslated region (3′-UTR) of the gene.
9. 2,174 of CGIs are within Introns (7.7 %), twice as many as the mouse. 104 of these are “copies” of a CGI, carried by jumping genes (retrotransposons) from another genomic site in the human genome
This was a surprise to me. 2,174 of the 37,000+ CGIs in the human genome are in introns and 2,033 are unique to humans, when these intronic CGIs are compared to mouse CGIs. In percentages, only 3.5% of mouse CGIs are found in introns, whereas 7.7% of human CGIs are found in introns. This means that the epigenetic regulation of genes in humans has evolved significantly to include regulatory regions in introns that can be silenced or amplified by DNA methylation. This is a major discovery about about “intron CGI evolution” that was just made in 2014 (see ref). Even more fascinating was the discovery that 104 of these newly evolved intronic CGIs found in the human genome were copies of another specific CGI in the human genome, clearly establishing strong circumstantial evidence that a retrotransposon “copied and pasted” the CGI into the new (intron) site. Specifically, the CGI in intron 2 of the Retinoblastoma (Rb) gene, found on chromosome 13, is a “carbon copy” of the CGI found in the gene on chromosome 9 called PPP1R26.
Reference: 2014 Evolutionary Origin and Methylation Status of Human Intronic CpG Islands that Are Not Present in Mouse
10. Some CGIs found in introns are imprinted (I.e. Epigenetic imprinting of intron CpGs)
This was also a fascinating surprise to me as well. So far, only a sample of specific intronic CGIs have been studied. One that has garnered a lot of attention is the CGI in intron 2 of the Retinoblastoma gene (Rb). Specifically the Rb gene has a CGI in intron 2 that is “differentially methylated.” In one allele of Rb, this intronic CGI is hypermethylated and in the other allele, the intronic CGI is unmethylated.
Reference: 2014 Evolutionary Origin and Methylation Status of Human Intronic CpG Islands that Are Not Present in Mouse
11. 10% of Transposable Elements (TEs) also have CpG islands
Some retrotransposons have CpG islands that are located in the 5′-UTR or 3′-UTR region of the transposable element (TE). Initially, only 16 human-specific retrotransposon copies were identified that had CpG islands in them. With time, more and more CoG islands were identified in TEs and now the experts believe that 1,319 TEs (approximately 10%) have CpGs. Unlike CpG islands in the rest of the human genome (where 60-70% of the cytosines are unmethylated), 68% of the retrocopy cytosine residues in the CpG islands are methylated. Some of these TEs are in gene deserts, but some are within the introns of genes, such as the Rb gene. Recently, they have proven that these TEs are actively transcribed in humans, especially in old age, when epigenetic repression of TEs fails.
References:
2016 Genome-wide methylation analysis of retrocopy-associated CpG islands and their genomic environment
“Gene duplication by retrotransposition, i.e., the reverse transcription of an mRNA and integration of the cDNA into the genome, is an important mechanism in evolution. Based on whole-genome bisulfite sequencing of monocyte DNA, we have investigated the methylation state of all CpG islands (CGIs) associated with a retrocopy (n = 1,319), their genomic environment, as well as the CGIs associated with the ancestral genes. Approximately 10% of retrocopies are associated with a CGI. Whereas almost all CGIs of the human genome are unmethylated, 68% of the CGIs associated with a retrocopy are methylated. In retrocopies resulting from multiple retrotranspositions of the same ancestral gene, the methylation state of the CGI often differs. There is a strong positive correlation between the methylation state of the CGI/retrocopy and their genomic environment, suggesting that the methylation state of the integration site determined the methylation state of the CGI/retrocopy, or that methylation of the retrocopy by a host defense mechanism has spread into the adjacent regions. Only a minor fraction of CGI/retrocopies (n = 195) has intermediate methylation levels. Among these, the previously reported CGI/retrocopy in intron 2 of the RB1 gene (PPP1R26P1) as well as the CGI associated with the retrocopy RPS2P32 identified in this study carry a maternal methylation imprint. In conclusion, these findings shed light on the evolutionary dynamics and constraints of DNA methylation.”
SECTION E EPIGENETICS AND DNA METHYLATION AND DISEASES
1. Epigenetic plays a major role in the pathogenesis of Type II Diabetes
There is now strong evidence that at least one important gene is epigenetically silenced in the pancreas with Type II diabetes. Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (aka PGC-1a) is a master regulator of mitochondrial gene expression. It has a CpG island within its promoter and in the pancreas, this CGI displays increased DNA methylation. Thus the PGC-1a gene is epigenetically down regulated in Type II diabetes and may explain both the obesity side of Type II diabetes, as well as the reduction in insulin secretion in the late phases of Type II diabetes. GCI methylation is not the only epigenetic mechanism seen in Type II diabetes, however. Histone H3K9 trimethylation and microRNA silencing of genes via the inhibition of mRNA translation has also been found to occur with Type II Diabetes (miR-375, miR-9, miR-192, miR-143, and others).
Reference: 2009 Epigenetic and microRNA-mediated regulation in diabetes
2. Epigenetic plays a major role in the pathogenesis of Cancer
Much of what we know about pathologic epigenetic phenomena has come from cancer research. Four abnormal DNA methylation patterns are seen in cancer cells, when compared to normal cells. Thus, it appears that cancer cells have learned to hijack the machinery of methylation/demethylatiom in the interest of their own survival.
- In normal cells, repetitive DNA (transposable elements) is hypermethylated, whereas in cancer cells, Yes they are hypomethylated.
- In normal cells, CpG islands found in promoters are hypomethylated, whereas in cancer the CpG islands are hypermethylated (especially tumor suppressor genes). Examples of this in cancer include epigenetic silencing of the following genes: MLH1, CDKN2A (p16INK4a), MGMT, DAPK, and CDH1.
- In normal cells, CpG island shores found upstream from the CGIs are not methylated, whereas in cancer cells, the CpG island shores are methylated.
- In normal cells, the gene bodies are methylated, whereas in cancer cells, the gene bodies are hypomethylated, which leads to the initiation of transcription at the wrong sites (not at the transcription start site).
Here is an illustration of these four4 differences between normal cell and cancer cell DNA methylation:
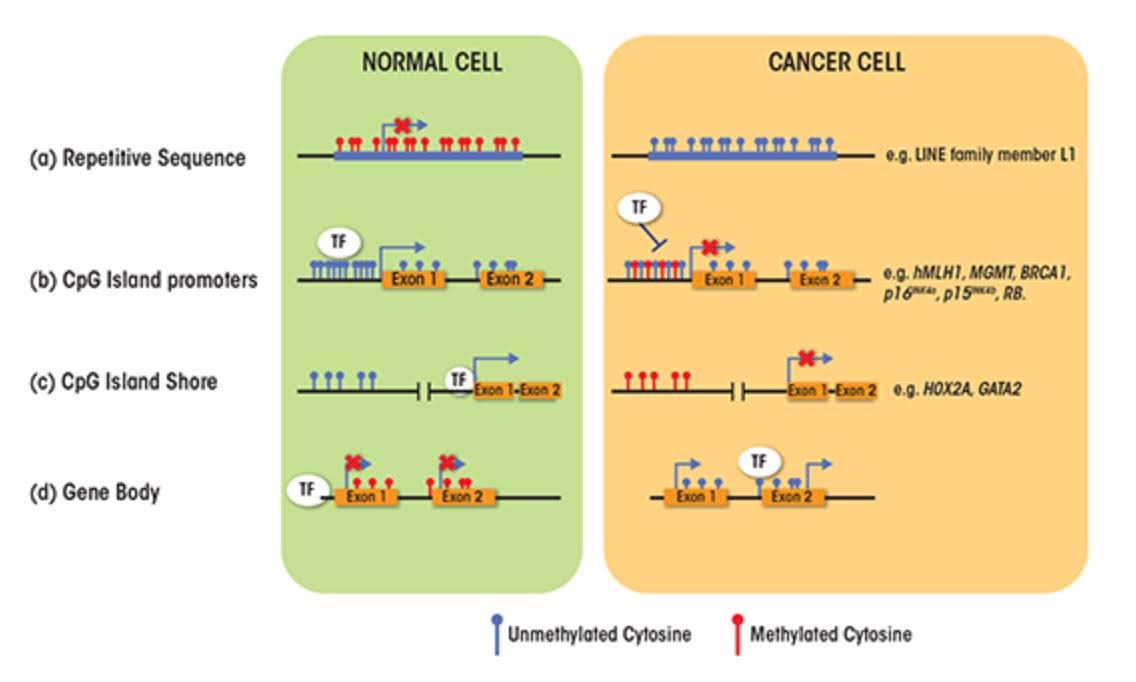
Reference and image source: Alcohol, DNA Methylation, and Cancer
Specifically, gene silencing (without any DNA mutation) can silence a tumor suppressor gene and trigger carcinogenesis. This can occur with both alleles of a specific tumor suppressor gene (i.e. purely epigenetic silencing) or it can occur where one allele is silenced by CGI methylation and the other allele may be mutated (inherited copy of a mutated gene). This 2nd scenario is referred to as “Loss of Heterozygosity” (LOH) and is thought to be the main way that tumor suppressor function is lost in cancer (both copies of a tumor suppressor must be silenced or mutated for cancer to occur, whereas only one copy of an oncogene must be amplified for cancer to occur).
Here is an illustration of how deletions, mutations, and epigenetic Loss of Heterozygosity can all result in the loss of tumor suppressor gene function in cancer:
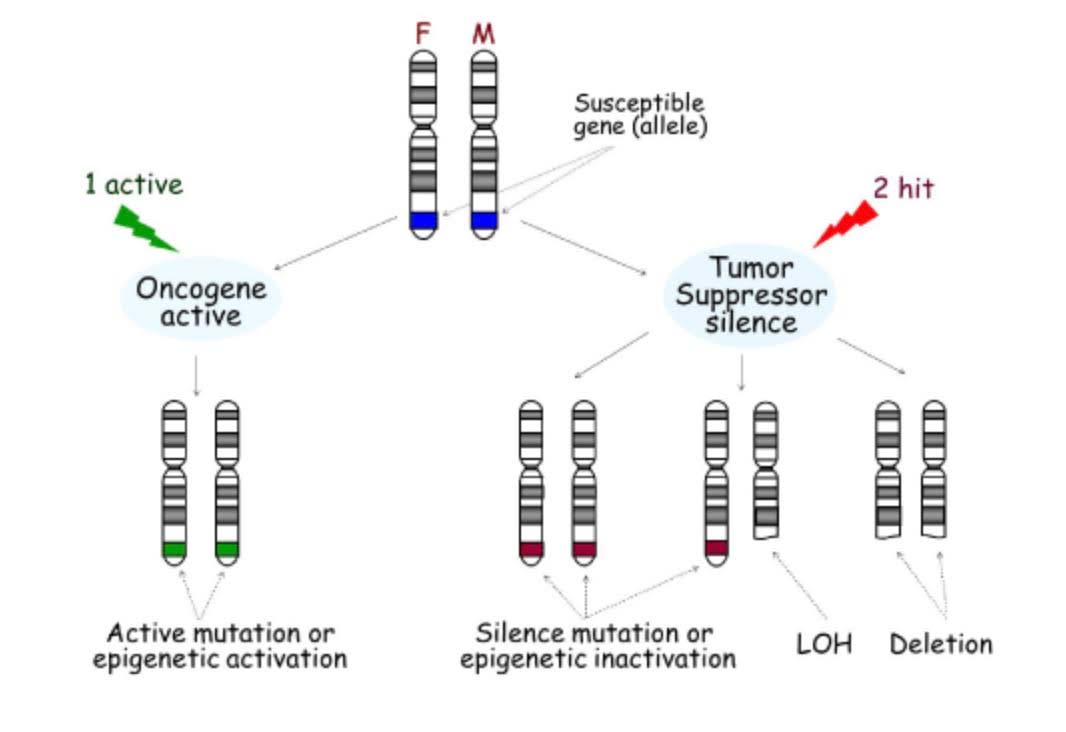
Reference and image source: 2014 Genetics and Epigenetics in Tumorigenesis: Acting Separately or Linked? “Development of cancer can occur through two main mechanisms. One “active,” usually an active mutation or epigenetic activation in oncogenes in either the father or mother (seen in green), is necessary for the activation of the oncogene, leading to tumorigenesis. On the other hand, tumor suppressors must be silenced by two “hits,” including silence mutation, epigenetic inactivation, Loss of Heterozygosity (LOH), or deletion (seen in red). F: Father; M: Mother”
For instance in breast cancer cell lines, between 5% and 14% of CGIs are found to be hypermethylated in a genome-wide study of CpG islands in breast cancer. The primary molecular mechanism behind this aberrent DNA methylation appears to be excessive estrogen exposure. One of the most common genes to become epigenetically silenced in breast cancer is the BRCA1 gene. The BRCA1 gene has a CGI within its promoter and it often becomes hypermethylated. In persons already carrying one bad copy of the BRCA1 gene (mutation carriers), this results in the development of breast cancer by “Loss of Heterozygosity”.
Another gene that is often hypermethylated at the CGI found in its promoter is the FHIT tumor suppressor gene, which becomes hypermethyated in breast cancer. Interestingly, one of the mechanisms by which alcohol consumption plays a role in breast cancer is via epigenetics. One drink of alcohol per day only increases the risk of breast cancer by 4%, but drinking 3 or more glasses of alcohol per day increases the risk of breast cancer by 40-50%. In 2010, Christenson and colleagues looked at the methylation profiles of 1,413 CpG sites and found a trend towards lower DNA methylation with increasing alcohol intake. Gene specific differential DNA methylation has been documented with alcohol-related breast cancer with the p16 gene becoming hypomethylated whereas the ER-alpha gene and E-cadherin genes become hypermethylated. Mutations in the MTHFR gene modulates the risk of alcohol-related breast cancer. Those with the “TT genotype” of the C677T MTHFR gene variant have a higher risk of breast cancer with alcohol intake.
References:
Alcohol, DNA Methylation, and Cancer
2014 Genetics and Epigenetics in Tumorigenesis: Acting Separately or Linked?
Prostate cancer is another cancer where epigenetics plays a major role. During the early phases of prostate cancer development, the CpG island in the ER-beta gene becomes hypermethylated, leading to the epigenetic silencing of ER-b. After the cancer metastasizes, however, the expression of the ER-b gene re-appears due to demethylation.
Reference: 2014 Genetics and Epigenetics in Tumorigenesis: Acting Separately or Linked?
A specific subset of cancers seem to have a tendency to undergo CpG methylation. These cancers are referred to as “CpG island methylator phenotype”, or CIMP. These cancers are clinically and etiologically distinct from most cancers and are characterized by “epigenetic instability”. The 1st cancer found to be a CIMP was a subset of colon cancers, characterized by microsatellite instability (MSI). These cancers had an increased frequency of promoter CGI methylation in multiple genes, most commonly the CDKN2A gene (which encodes for the p16INK4a protein and thrombosponsin 1) and the MLH1 gene (which encodes for a mismatch repair enzyme). Another gene that are frequently found to have hypermethylated CGIs in the CIMP type of cancers includes the HPP1 gene.
Little by little, the molecular mechanisms of how the CpG island methylator phenotype epigenetic phenomena occurs has been unravelling. The epigenetic story of CIMP is clearly intertwined with the epigenetic story of aging. Specifically with aging, border methylation of CpG islands occurs. With multiple cell divisions, the border methylation “creeps” in and starts to methylate the CpG island itself. Thus “aging” precedes “cancer” in the epigenetic sequence of CpG island silencing by hypermethylation. Following is an illustration of this phenomena, showing the role of “aging” in triggering the CIMP subset of cancers.
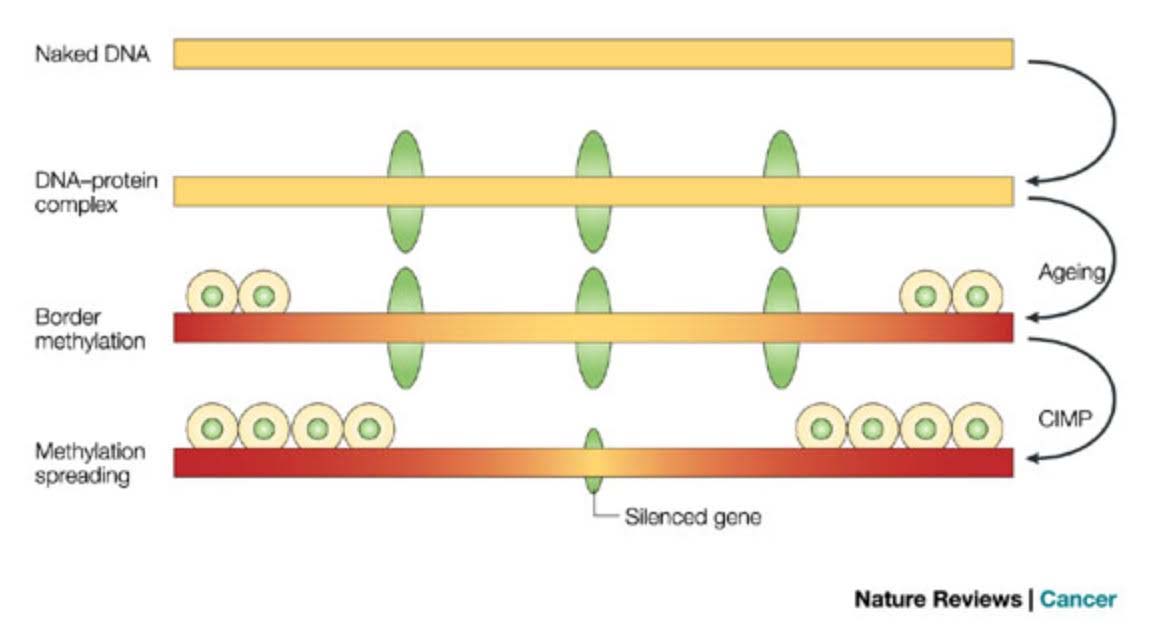
Image source: A model of hypermethylation in cancer
“Naked CpG island DNA (top) is unmethylated (yellow) and coated by proteins (green ovals) that protect against DNA methylation establishment and/or spreading. The nature of these proteins is unknown, but probably include transcription factors, co-activators or similar molecules. During repeated rounds of the stem-cell mobilization and replication that accompany ageing, DNA methyltransferases (circles) are recruited to the borders of some CpG islands, depositing methyl groups (red) and creating methylation pressure for these islands. The nature of this initial recruitment is unknown but is probably related to repetitive DNA sequences and/or retrotransposons. The balance of methylation pressure (circles) and methylation protection (ovals) is disrupted in the CpG island methylator phenotype (CIMP), resulting in the spread of methylation into the transcription start area and the triggering of the silencing cascade. As discussed in the text, the disruption of this balance will probably be achieved through the loss of protective proteins (as indicated in the bottom panel), which could occur by mutations that inactivate these proteins or the loss of expression by other mechanisms such as transcription factor loss or histone modifications. Theoretically, this balance could also be disrupted by overactive de novo methylation pressure (circles), for example by activating mutations in DNA methyltransferases.”
References:
1999 Methylation Profiling of CpG Islands in Human Breast Cancer Cells
2004 CpG island methylator phenotype in cancer
3. The Six Major Genome-wide changes in DNA methylation that occur with Cancer
Although there are clearly major differences between aging and cancer, there are more similarities than there are differences. Because of the enormous amount of funding and research that has been done on cancer, compared to the small amount of funding and research on aging, there is much we can learn from cancer researchers about DNA methylation that may also occur in aging. With cancer, there are 6 common features of differential DNA methylation that occur – 1. repetitive sequence demethylation, 2. hypomethylation of specific promoters, 3. hypermethylation of CpG islands in specific promoters, 4. loss of imprinting of imprinted genes and the paternal X chromosome, 5. abnormal methylation of CpG island shores, and 6. demethylation of gene bodies. Four of these six changes are depicted below. These same changes likely occur with aging too.
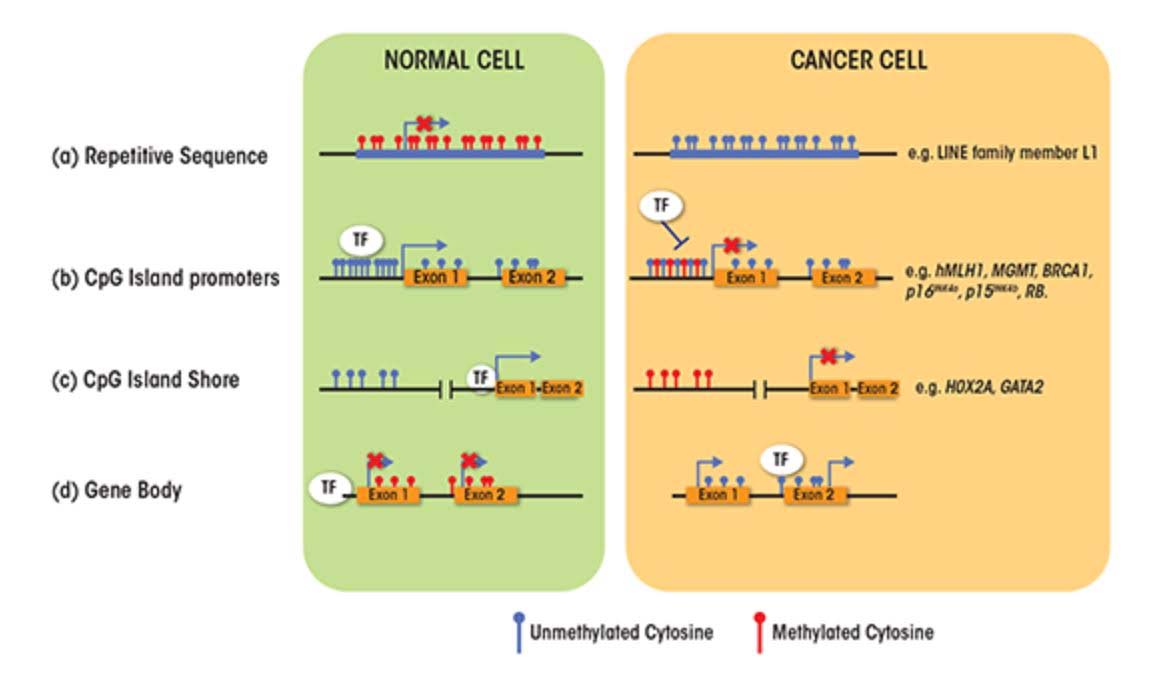
Reference and image source:: Alcohol, DNA Methylation, and Cancer
a) Loss of DNA methylation in repetitive DNA sequences => chromosomal/genomic instability, telomere attrition, and aneuploidy
Repetitive sequences is a type of DNA that used to be called “junk”, but now it is known to be actively transcribed with aging and with disease. Silencing this type of DNA has been shown to prevent or delay the onset of aging and many disease, but is an enormous task since over 50% of the human genome is made of repetitive sequences. One of the major ways that repetitive DNA sequences are silenced is by DNA methylation of cytosine in CpG dinucleotides. During the S-phase of the cell cycle, when DNA is replicated (copied), the newly synthesized strand of DNA must be methylated, based on the methyl cytosine pattern of the original DNA strand. The enzyme DNA methyltransferase 1 (DNMT1) performs this task. Unfortunately this gene is down-regulated with aging. Furthermore, it cannot “do its job” if there is a methyl donor deficiency, such as what happens with folate deficiency, B12 deficiency, or with MTHFR gene mutations. Even in cells that are no longer dividing, there are two DNA methyltransfereases that can maintain normal DNA methylation patterns – DNMT3a and DNMT3b. Although DNMT3b increases with aging, DNMT3a decreases with aging. Thus the specific reasons for this opposing change in DNMT3 gene expression is not entirely clear now. What is clear is that Alu repeats, the most common repetitive sequence in the human genome (and is primate-exclusive, or primate-specific), undergoes progressive loss of DNA methylation with aging. Another common repetitive sequence that looses its DNA methylation with aging is the Human Endogenous Retroviral genes (HERV), of which there are over 100,000 copies in the human genome. With cancer, as many as 20-60% of the methylated cytosines in CpG dinucleotides undergo demethylation. With aging, this occurs as well, but at a slower rate. With alcohol abuse, repetitive DNA loss of CpG methylation occurs at an accelerated rate. This explains one reason why alcohol accelerates age-related diseases such as dementia and depression, as well as why alcohol accelerates or plays a causal role in carcinogenesis.
b) CpG Island promoters become hypermethylated with aging –aka Type A methylator pattern
c) CpG Island shores become hypermethylated with aging
d) Gene bodies lose their DNA CpG methylation with aging and disease
SECTION F – ON TO INTERVENTIONS?
(Final Comments by Vince Giuliano)
The viewpoint of this blog entry – that of DNA methylation -does not tell everything about aging, But it opens the door for specific suggestions of what might be done to maintain health in aging and perhaps even to lead to longer healthy lives. Here is a recap of how I see that situation, in largely non-technical terms.
There are two major observed DNA methylation trends observed with advancing age: 1. Global hypomethylation of CGIs in non-promoter sites. This leads to expression of many genes we would not like to see over-expressed such as oncogenes and pro- inflammatory genes, and it likely leads to over expression of transposable elements. The consequence can be dysregulation of protein expression leading to diseases of old age, essentially all of which are inflammatory in origin. And, 2. Selective hypermethylation of CGIs of promoters for health-maintaining genes, such as ones that limit inflammation and carcinogenic processes. This turns off genes that produce protective proteins, again opening the door for many diseases of old age.
We can do some things to possibly mitigate both trends. Here are a few ideas in a nutshell, ones I personally pursue.
1, Global hypomethylation: In Item 12 of Section B and in Item 2 of Section C above we discussed the role of cortisol and dysregulation of circadian rhythms in inducing global DNA hypomethylation. We talked about how cortisol can induce expression of TET enzymes that induce hypomethylation. This suggests that with aging certain hormone supplements might be useful, namely melatonin at bedtime to help stablilize the circadian cycle and DHEA supplementation which antagonizes cortisol expression. The ratio of DHEA and cortisol appears to be quite important. Also suggested is possible supplementation with pregnenalone, a “mother” hormone for both cortisol and DHEA. And I believe long regular sleep can help offset the circadian dysregulation often experienced by people in their 80s and above.
Failure to maintain the silence of repetitive sequences of DNA can also by a consequence of global hypomethylation. See Item 7 in Section B and Item 3a in Section E, As pointed out, the gene for the DNA methyltransferase 1 (DNMT1) which works during the S-phase of the cell cycle is down-regulated with aging and its working is dependent on the availability of an adequate supply of methyl doner molecules. Therefore, supplementation with Vitamin B12 and folic acid might be a good strategy
2. Selective hypermethylation: In Items 10 and 11 of Section B we explained how Polycomb protein Complex target genes become hypermethylated in both aging and cancer the link being inflammation. Item 12 in Section B discusses how chronic stress can lead to accelerated aging via age-assocated DNA methylation. We discussed this further in Item 1 of Section C and specifically talked about how the inflammatory cytokine IL-6 “drives” age-related epigenetic hypermethylation, As I have often written, we know that chronic inflammation is at the heart of essentially all killer diseases related to aging. Therefore this finding with respect to selective DNA hypermethylation lends additional weight to strategies for control of chronic inflammation, The topic of chronic inflammation is very complex. While inflammation is an essential natural process, chronic inflammation can have many possible causes. And there are many possible strategies for dealing with it. Jim Watson and I are currently planning a series of blog entries on chronic inflammation, and they should soon begin to appear. What I mention here is only a few points that have been highly important for me both professionally and personally: a) that strategies for controlling age-related inflammation do exist and can be highly effective, b) I have come to believe supplementation with certain traditional well-studied herbs together with observing some simple dietary and lifestyle rules can be a highly effective approach for controlling several pernicious forms of chronic inflammation, and c) that with colleagues I have developed a liposomal (high bioavailability) preparation of four powerful anti-inflammatory herbal extracts that I and others have been consuming for three years now, and this should soon should be available commercially.

{kind=link}
Pingback: Inflammation Part 2: The Tale of Three Stress Sensors and their Interactions: 1)Inflammation, 2)Genomic Instability (p53), and 3)Oxidative stress (Nrf2) | AGINGSCIENCES™ – Anti-Aging Firewalls™
Pingback: ON AGING - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™
Pingback: Two faces of Life Extension - AGINGSCIENCES™ - Anti-Aging Firewalls™AGINGSCIENCES™ – Anti-Aging Firewalls™